Code
!pip install dldna[colab] # in Colab
# !pip install dldna[all] # in your local
%load_ext autoreload
%autoreload 2 ![]()
“Einfachheit ist die ultimative Eleganz.” - Leonardo da Vinci
Deep-Learning-Modelle verfügen über eine starke Fähigkeit, komplexe Funktionen durch zahlreiche Parameter auszudrücken. Diese Fähigkeit kann jedoch manchmal wie ein Doppelschwert sein. Wenn das Modell sich zu sehr an die Trainingsdaten anpasst, tritt die Überanpassung (overfitting) auf, bei der die Vorhersageleistung für neue Daten tatsächlich sinkt.
Nachdem 1986 der Rückpropagationsalgorithmus wiederentdeckt wurde, war Überanpassung eine ständige Herausforderung für Deep-Learning-Forscher. Anfangs reagierte man auf Überanpassung, indem man die Modellgröße reduzierte oder mehr Trainingsdaten hinzufügte. Diese Methoden hatten jedoch Grenzen, da sie die Ausdrucksfähigkeit des Modells einschränkten oder durch Schwierigkeiten bei der Datensammlung beeinträchtigt wurden. 2012 markierte das Erscheinen von AlexNet eine neue Ära für Deep Learning, aber es unterstrich gleichzeitig die Dringlichkeit des Überanpassungsproblems. AlexNet verfügte über viel mehr Parameter als frühere Modelle und damit auch um einiges größere Überanpassungsrisiken. Mit der exponentiellen Vergrößerung von Deep-Learning-Modellen wurde das Überanpassungsproblem zum zentralen Thema in der Forschung.
In diesem Kapitel werden wir die Essenz der Überanpassung verstehen und verschiedene Methoden zur Lösung dieses Problems betrachten, die im Laufe der Zeit entwickelt wurden. Wie Entdecker, die unbekanntes Territorium erforschen und Karten erstellen, haben Deep-Learning-Forscher ständig neue Wege erforscht und verbessert, um das Problem der Überanpassung zu bewältigen.
Die Überanpassung wurde erstmals 1670 in William Hopkins’ Schrift erwähnt, aber im modernen Sinne begann sie 1935 im Quarterly Review of Biology mit der Bemerkung “Eine sechsfache Analyse von 13 Beobachtungen sieht wie Überanpassung aus”. Sie wurde ab den 1950er Jahren in der Statistik intensiver untersucht, insbesondere in der Arbeit “Tests of Fit in Time Series” aus dem Jahr 1952 im Kontext der Zeitreihenanalyse.
Das Überanpassungsproblem im Deep Learning nahm eine neue Dimension an, als AlexNet 2012 erschien. AlexNet war ein großes neuronales Netz mit etwa 60 Millionen Parametern und überragte damit die vorherigen Modelle in der Größenordnung. Mit der exponentiellen Vergrößerung von Deep-Learning-Modellen wurde das Überanpassungsproblem immer dringlicher. Zum Beispiel verfügen moderne große Sprachmodelle (LLMs) über Hunderte von Milliarden Parametern, wodurch die Prävention von Überanpassung zu einem zentralen Designziel geworden ist.
Um dieser Herausforderung zu begegnen, wurden innovative Lösungen wie Dropout (2014), Batch-Normalisierung (2015) vorgeschlagen, und in jüngerer Zeit werden fortschrittlichere Methoden zur Erkennung und Verhinderung von Überanpassung durch die Nutzung von Trainingsverläufen (2024) erforscht. Besonders bei großen Modellen werden verschiedene Strategien kombiniert verwendet, von traditionellen Ansätzen wie early stopping bis hin zu modernen Techniken wie Ensemble-Lernen und Datenverstärkung.
Lasst uns ein einfaches Beispiel betrachten, um das Phänomen der Überanpassung intuitiv zu verstehen. Wir wenden Polynome unterschiedlichen Grades auf Daten an, die eine sinusförmige Funktion mit Rauschen enthalten.
!pip install dldna[colab] # in Colab
# !pip install dldna[all] # in your local
%load_ext autoreload
%autoreload 2import numpy as np
import seaborn as sns
# Noisy sin graph
def real_func(x):
y = np.sin(x) + np.random.uniform(-0.2, 0.2, len(x))
return y
# Create x data from 40 to 320 degrees. Use a step value to avoid making it too dense.
x = np.array([np.pi/180 * i for i in range(40, 320, 4)])
y = real_func(x)
import seaborn as sns
sns.scatterplot(x=x, y=y, label='real function')
# Plot with 1st, 3rd, and 21th degree polynomials.
for deg in [1, 3, 21]:
# Get the coefficients for the corresponding degree using polyfit, and create the estimated function using poly1d.
params = np.polyfit(x, y, deg) # Get the parameter values
# print(f" {deg} params = {params}")
p = np.poly1d(params) # Get the line function
sns.lineplot(x=x, y=p(x), color=f"C{deg}", label=f"deg = {deg}")The autoreload extension is already loaded. To reload it, use:
%reload_ext autoreload/tmp/ipykernel_1362795/2136320363.py:25: RankWarning: Polyfit may be poorly conditioned
params = np.polyfit(x, y, deg) # Get the parameter values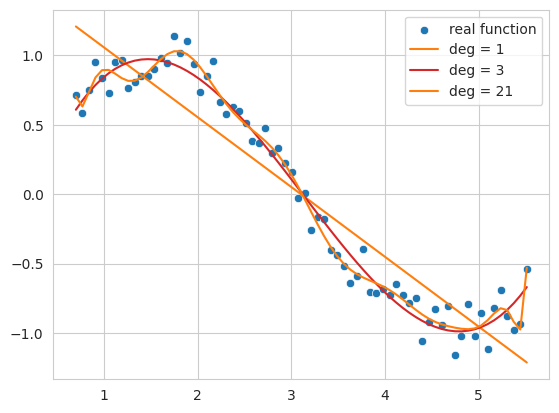
Der folgende Code erstellt Daten einer Sinusfunktion mit Rauschen und passt (fitting) diese Daten mit Polynomen 1. Ordnung, 3. Ordnung und 21. Ordnung an.
Polynom 1. Ordnung (deg = 1): Es folgt nicht dem allgemeinen Trend der Daten und zeigt eine einfache lineare Form. Dies zeigt einen Zustand von Underfitting, bei dem das Modell die Komplexität der Daten nicht ausreichend darstellt.
Polynom 3. Ordnung (deg = 3): Es erfasst das grundlegende Muster der Daten ziemlich gut und zeigt eine glatte Kurve, die nicht stark durch Rauschen beeinflusst wird.
Polynom 21. Ordnung (deg = 21): Es folgt zu sehr dem Rauschen in den Trainingsdaten und zeigt einen Zustand von Overfitting, bei dem das Modell übermäßig an die Trainingsdaten angepasst ist.
So tritt Underfitting auf, wenn die Komplexität des Modells (hier der Grad des Polynoms) zu niedrig ist, und Overfitting, wenn sie zu hoch ist. Letztendlich suchen wir ein Modell, das nicht nur die Trainingsdaten, sondern auch neue Daten gut generalisieren kann, also eine Näherungsfunktion, die dem tatsächlichen Sinus am nächsten kommt.
Overfitting tritt auf, wenn die Komplexität (Kapazität) des Modells im Verhältnis zur Menge der Trainingsdaten relativ groß ist. Neuronale Netze haben aufgrund ihrer vielen Parameter und hohen Ausdrucksstärke eine besondere Anfälligkeit für Overfitting. Wenn die Trainingsdaten knapp sind oder viel Rauschen enthalten, kann Overfitting ebenfalls auftreten. Overfitting zeigt sich durch folgende Merkmale:
Letztendlich zeigt ein über angepasstes Modell hohe Leistung auf den Trainingsdaten, fällt aber bei neuen Daten in Bezug auf Vorhersageleistung zurück. Um Overfitting zu vermeiden, werden wir uns im Folgenden mit verschiedenen Techniken wie L1/L2-Regularisierung, Dropout und Batch-Normalisierung näher beschäftigen.
Herausforderung: Wie kann die Generalisierungsleistung verbessert werden, während gleichzeitig die Komplexität des Modells effektiv gesteuert wird?
Überlegungen der Forscher: Das Verkleinern der Modellgröße, um das Overfitting zu verhindern, kann die Ausdrucksfähigkeit einschränken und eine einfache Erhöhung der Trainingsdatenmenge ist nicht immer möglich. Es war notwendig, Methoden zu finden, die durch Einschränkungen an der Struktur oder im Lernprozess des Modells übermäßige Optimierung für die Trainingsdaten verhindern und die Vorhersageleistung für neue Daten verbessern.
Typische Regularisierungsverfahren in neuronalen Netzen sind die L1- und L2-Regularisierung. L1 steht für Lasso, L2 für Ridge-Regression (oder lineare Regression mit diesen Techniken).
Sie werden auch als Ridge-Regression und Lasso-Regression bezeichnet, wobei jede eine bestimmte Strafterm einführt, um die Bewegung der Parameter zu begrenzen. Die Unterschiede zwischen den beiden Methoden können in folgender Tabelle zusammengefasst werden:
| Merkmal | Ridge-Regression (Ridge Regression) | Lasso-Regression (Lasso Regression) |
|---|---|---|
| Strafarten | L2-Strafe anwenden. Der Strafterm ist das Produkt des Quadrats der Parameter und des Alpha-Werts. | L1-Strafe anwenden. Der Strafterm ist das Produkt der absoluten Werte der Parameter und des Alpha-Werts. |
| Einfluss auf die Parameter | Dämpft große Parameter, sodass sie nahe bei 0 liegen (aber nie exakt 0). | Bei hohen Alpha-Werten kann es dazu führen, dass einige Parameterwerte exakt 0 werden, was zu einem vereinfachten Modell führt. |
| Gesamt-Einfluss | Alle Parameter bleiben erhalten; auch solche mit geringerem Einfluss bleiben bestehen. | Nur relevante Parameter bleiben übrig und die Methode besitzt selektive Eigenschaften, was dazu beiträgt, komplexe Modelle einfacher zu beschreiben. |
| Optimierungseigenschaften | Weniger empfindlich gegenüber idealen Werten im Vergleich zu Lasso. | Aufgrund des absoluten Wertes der Strafterme ist die Methode empfindlicher gegenüber idealen Werten. |
Die mathematischen Darstellungen sehen wie folgt aus:
Ridge-Zielfunktion (Ridge Regression Objective Function)
“Modifizierte Ridge-Zielfunktion” = (unmodifizierte lineare Regressionsfunktion) + \(\alpha \cdot \sum (\text{Parameter})^2\)
\(f_{\beta} = \sum_{i=1}^{M} (y_i - \hat{y}_i)^2 + \alpha \sum_{j} \beta_{j}^2\)
Hierbei ist \(\beta\) der zu bestimmende Parameter-Vektor (Gewichte). \(\alpha \sum_{j} \beta_{j}^2\) wird als Strafterm oder Regularisierungsterm bezeichnet. \(\alpha\) ist ein Hyperparameter, der die Stärke des Regularisierungsterms steuert. Die Formel zur Bestimmung der Parameter lautet:
\(\beta = \underset{\beta}{\operatorname{argmin}} \left( \sum_{i=1}^{M} (y_i - \hat{y}_i)^2 + \alpha \sum_{j} \beta_{j}^2 \right)\)
Lasso-Zielfunktion (Lasso Regression Objective Function)
“Modifizierte Lasso-Zielfunktion” = (unmodifizierte lineare Regressionsfunktion) + $ || $
\(f_{\beta} = \sum_{i=1}^{M} (y_i - \hat{y}_i)^2 + \alpha \sum_{j} |\beta_{j}|\) \(\beta = \underset{\beta}{\operatorname{argmin}} \left( \sum_{i=1}^{M} (y_i - \hat{y}_i)^2 + \alpha \sum_{j} |\beta_j| \right)\)
Die Summe der Quadrate der Parameter als Strafterm zu verwenden, wird in neuronalen Netzen häufig als Gewichtsverfall (weight decay) bezeichnet. Wir werden untersuchen, wie sich die Verwendung von Ridge-Regression (L2) im Vergleich zur einfachen linearen Regression unterscheidet. Dafür verwenden wir ein Modell, das in sklearn implementiert ist. Dazu müssen wir die Eingabedaten x so verarbeiten, dass sie um die entsprechende Anzahl von Dimensionen erweitert werden. Wir werden dies mit der folgenden einfachen Utility-Funktion tun.
def get_x_powered(x, p=1):
size = len(x)
# The shape of the created x will be (data size, degree)
new_x = np.zeros((size, p))
for s in range(len(x)): # Iterate over data size
for d in range(1, p+1): # Iterate over degrees
new_x[s][d-1] = x[s]**d # Raise x to the power of the degree.
return new_x
# Let's take a quick look at how it works.
deg = 3
x = np.array([np.pi/180 * i for i in range(20, 35, 5)])
y = real_func(x) # real_func는 이전 코드에 정의되어 있다고 가정
print(f"x = {x}")
new_x = get_x_powered(x, p=deg)
print(f"new_x = {new_x}")x = [0.34906585 0.43633231 0.52359878]
new_x = [[0.34906585 0.12184697 0.04253262]
[0.43633231 0.19038589 0.08307151]
[0.52359878 0.27415568 0.14354758]]Da es sich um eine dritte Ordnung handelt, erhöhen sich die \(x\)-Werte zu \(x^2, x^3\). Zum Beispiel 0.3490, 0.1218 (Quadrat von 0.3490), 0.04253 (Kubik von 0.3490). Wenn es sich um eine zehnte Ordnung handelt, werden die Daten bis \(x^{10}\) generiert. Der Alphawert des Strafterms kann Werte von null bis unendlich annehmen. Je größer der Alphawert ist, desto stärker ist die Regularisierung. Wir werden den Grad auf 13 festlegen und die lineare Regression mit Ridge-Regression bei unterschiedlichen Alphawerten vergleichen.
import numpy as np
from sklearn.linear_model import Ridge
import matplotlib.pyplot as plt
import seaborn as sns
from sklearn.preprocessing import PolynomialFeatures
from sklearn.pipeline import make_pipeline
# Create a noisy sine wave (increased noise)
def real_func(x):
return np.sin(x) + np.random.normal(0, 0.4, len(x)) # Increased noise
# Create x data (narrower range)
x = np.array([np.pi / 180 * i for i in range(40, 280, 8)]) # Narrower range, larger step
y = real_func(x)
# Degree of the polynomial
deg = 10
# List of alpha values to compare (adjusted)
alpha_list = [0.0, 0.1, 10] # Adjusted alpha values
cols = len(alpha_list)
fig, axes_list = plt.subplots(1, cols, figsize=(20, 5)) # Adjusted figure size
for i, alpha in enumerate(alpha_list):
axes = axes_list[i]
# Plot the original data
sns.scatterplot(ax=axes, x=x, y=y, label='real function', s=50) # Increased marker size
# Plot linear regression
params = np.polyfit(x, y, deg)
p = np.poly1d(params)
sns.lineplot(ax=axes, x=x, y=p(x), label=f"LR deg = {deg}")
# Ridge regression (using Pipeline, solver='auto')
model = make_pipeline(PolynomialFeatures(degree=deg), Ridge(alpha=alpha, solver='auto'))
model.fit(x.reshape(-1, 1), y) # Reshape x for pipeline
y_pred = model.predict(x.reshape(-1, 1)) # Reshape x for prediction
sns.lineplot(ax=axes, x=x, y=y_pred, label=f"Ridge alpha={alpha:0.1e} deg={deg}")
axes.set_title(f"Alpha = {alpha:0.1e}")
axes.set_ylim(-1.5, 1.5) # Limit y-axis range
axes.legend()
plt.tight_layout()
plt.show()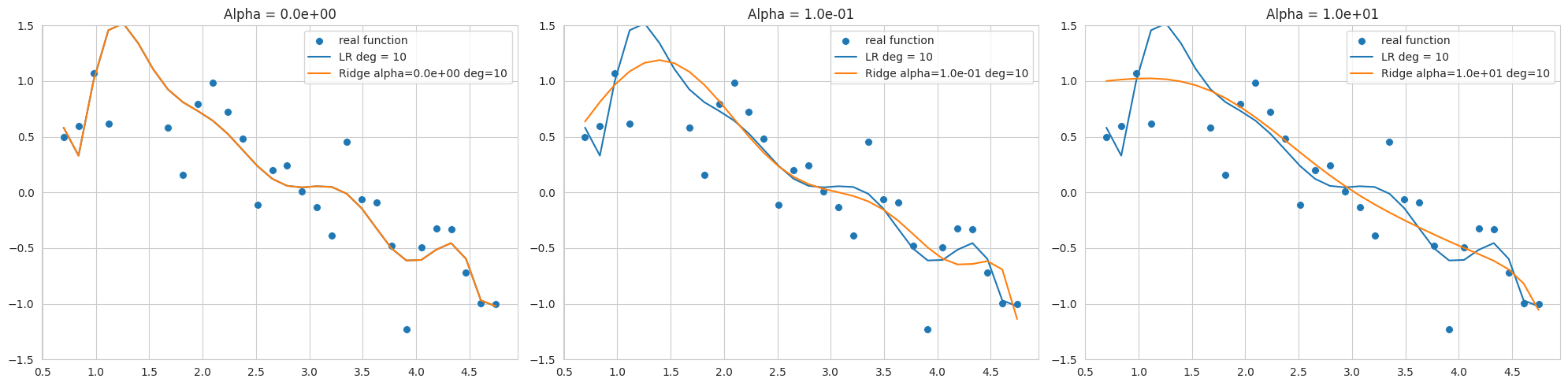
Das obige Diagramm zeigt die Ergebnisse des Fittings von Sinusfunktionsdaten mit hinzugefügtem Rauschen unter Verwendung eines Polynoms 10. Grades, wobei Ridge-Regression mit unterschiedlichen alpha Werten (Regularisierungsstärke) dargestellt wird. Da der Datenbereich eng und das Rauschen hoch ist, kann Overfitting selbst bei niedrigen Graden leicht auftreten.
alpha=0 reduziert wird, jedoch bleibt es noch empfindlich gegenüber Rauschen und weicht vom Sinusfunktion ab.Durch eine geeignete Auswahl des alpha Wertes kann die Modellkomplexität gesteuert und die Verallgemeinerungsleistung verbessert werden. L2-Regularisierung ist nützlich, da sie die Gewichte nahe an Null hält und das Modell stabilisiert.
Das sklearn.linear_model.Ridge Modell kann je nach gewähltem solver unterschiedliche Optimierungsverfahren verwenden. Insbesondere bei engen Datenbereichen mit viel Rauschen, wie in diesem Beispiel, können 'svd' oder 'cholesky' Solver stabiler sein, so dass bei der Auswahl des Solvers Vorsicht geboten ist (im Code wird 'cholesky' verwendet).
PyTorch und Keras unterscheiden sich darin, wie sie L1- und L2-Regularisierung implementieren. Keras unterstützt die direkte Hinzufügung von Regularisierungstermen zu jeder Schicht (kernel_regularizer, bias_regularizer).
# In Keras, you can specify regularization when declaring a layer.
keras.layers.Dense(64, activation='relu',
kernel_regularizer=regularizers.l2(0.01),
input_shape=(784,))Im Gegensatz dazu wendet PyTorch L2-Regularisierung durch die Einstellung des Gewichtsverfalls (weight decay) im Optimierer (optimizer) an und implementiert L1-Regularisierung in der Regel über benutzerdefinierte Verlustfunktionen.
import torch.nn as nn
import torch
def custom_loss(outputs, targets, model, lambda_l1=0.01, lambda_l2=0.01,):
mse_loss = nn.MSELoss()(outputs, targets)
l1_loss = 0.
l2_loss = 0.
for param in model.parameters():
l1_loss += torch.sum(torch.abs(param)) # Take the absolute value of the parameters.
l2_loss += torch.sum(param ** 2) # Square the parameters.
total_loss = mse_loss + lambda_l1 * l1_loss + lambda_l2 * l2_loss # Add L1 and L2 penalty terms to the loss.
return total_loss
# Example usage within a training loop (not runnable as is)
# for inputs, targets in dataloader:
# # ... (rest of the training loop)
# loss = custom_loss(outputs, targets, model)
# loss.backward()
# ... (rest of the training loop)Wie im obigen Beispiel gezeigt, kann die custom_loss-Funktion definiert werden, um sowohl L1- als auch L2-Regularisierung anzuwenden. Allerdings wird in der Regel das weight_decay, das der L2-Regularisierung entspricht, im Optimizer festgelegt und verwendet. Bei den Optimizern Adam und SGD wird die Gewichtsverringerung jedoch etwas anders implementiert als die traditionelle L2-Regularisierung. Die traditionelle L2-Regularisierung fügt dem Verlustfunktionsterm das Quadrat der Parameter hinzu.
\(L_{n+1} = L_{n} + \frac{ \lambda }{2} \sum w^2\)
Die Ableitung davon nach den Parametern ergibt folgendes:
\(\frac{\partial L_{n+1}}{\partial w} = \frac{\partial L_{n}}{\partial w} +\lambda w\)
SGD und Adam implementieren dies, indem sie den Term \(\lambda w\) direkt zur Gradientenaddition verwenden. Der Code für chapter_05/optimizers/ SGD sieht wie folgt aus.
if self.weight_decay != 0:
grad = grad.add(p, alpha=self.weight_decay)Dieses Verfahren hat genau die gleiche Wirkung wie das Hinzufügen eines L2-Regulierungs TERMS zum Verlustfunktion, wenn es mit Momentum oder adaptiven Lernraten kombiniert wird.
Trennung von AdamW und Gewichtsabkling (Decoupled Weight Decay)
In dem 2017 auf der ICLR veröffentlichten Papier “Fixing Weight Decay Regularization in Adam” (https://arxiv.org/abs/1711.05101) wird das Problem angesprochen, dass die Gewichtsabkling im Adam-Optimierer anders als L2-Regulierung funktioniert, und der modifizierte AdamW-Optimierer vorgeschlagen wird. In AdamW wird die Gewichtsabkling von den Gradientenupdates getrennt und direkt im Schritt der Parameteraktualisierung angewendet. Der Code befindet sich in derselben Datei basic.py.
# PyTorch AdamW weght decay
if weight_decay != 0:
param.data.mul_(1 - lr * weight_decay)AdamW multipliziert die Parameterwerte mit 1 - lr * weight_decay.
Zusammenfassend lässt sich sagen, dass AdamWs Ansatz einer genauen Implementierung der L2-Regulierung näher kommt. Obwohl die Gewichtsabkling bei SGD und Adam oft als L2-Regulierung bezeichnet wird, liegt dies an historischen Gründen und ähnlichen Effekten; streng genommen ist es jedoch korrekter, sie als separate Regularisierungstechniken zu betrachten. AdamW klärt diese Unterschiede und bietet dadurch bessere Leistung.
Um den Einfluss von L1- und L2-Regularisierung auf das Lernen des Modells visuell zu verstehen, werden wir die in Kapitel 4 vorgestellte Technik zur Visualisierung der Verlustfläche (loss surface) anwenden. Wir vergleichen die Änderungen der Verlustfläche bei fehlender Regularisierung und bei Anwendung von L2-Regularisierung und beobachten die Verschiebung der optimalen Lösung in Abhängigkeit von der Regularisierungsstärke (weight_decay).
import sys
from dldna.chapter_05.visualization.loss_surface import xy_perturb_loss, hessian_eigenvectors, visualize_loss_surface
from dldna.chapter_04.utils.data import get_dataset, get_device
from dldna.chapter_04.utils.metrics import load_model
import torch
import torch.nn as nn
import numpy as np
import torch.utils.data as data_utils
from torch.utils.data import DataLoader
device = get_device() # Get the device (CPU or CUDA)
train_dataset, test_dataset = get_dataset() # Load the datasets.
act_name = "ReLU"
model_file = f"SimpleNetwork-{act_name}.pth"
small_dataset = data_utils.Subset(test_dataset, torch.arange(0, 256)) # Use a subset of the test dataset
data_loader = DataLoader(small_dataset, batch_size=256, shuffle=True) # Create a data loader
loss_func = nn.CrossEntropyLoss() # Define the loss function
# Load the trained model.
trained_model, _ = load_model(model_file=model_file, path="./tmp/opts/ReLU") # 4장의 load_model 사용
trained_model = trained_model.to(device) # Move the model to the device
top_n = 2 # Number of top eigenvalues/eigenvectors to compute
top_eigenvalues, top_eigenvectors = hessian_eigenvectors(model=trained_model, loss_func=loss_func, data_loader=data_loader, top_n=top_n, is_cuda=True) # 5장의 함수 사용
d_min ,d_max, d_num = -1, 1, 50 # Define the range and number of points for the grid
lambda1, lambda2 = np.linspace(d_min, d_max, d_num).astype(np.float32), np.linspace(d_min, d_max, d_num).astype(np.float32) # Create the grid of lambda values
x, y, z = xy_perturb_loss(model=trained_model, top_eigenvectors=top_eigenvectors, data_loader=data_loader, loss_func=loss_func, lambda1=lambda1, lambda2=lambda2, device=device) # 5장의 함수 사용xy_perturb_loss wird verwendet, um eine Näherungsfunktion zu erstellen. Danach werden (x,y) in diese Näherungsfunktion eingesetzt, um einen neuen z-Wert zu berechnen. Der Grund dafür ist, dass die Werte, die mit xy_perturb_loss berechnet wurden, wie im Kapitel 5 gezeigt, Konturlinien erzeugen, bei denen der Minimalwert leicht abweicht und der Optimierer an einem leicht unterschiedlichen Punkt konvergiert. Jetzt werden nicht alle Pfade, auf denen der Optimierer verläuft, dargestellt, sondern nur der letzte tiefste Punkt wird verglichen, wobei der Dämpfungsparameter weight_decay schrittweise erhöht wird.
import torch
import numpy as np
import torch.nn as nn
import torch.optim as optim # Import optim
import matplotlib.pyplot as plt
from torch.utils.data import DataLoader, Subset
# 5장, 4장 함수들 import
from dldna.chapter_05.visualization.loss_surface import (
hessian_eigenvectors,
xy_perturb_loss,
visualize_loss_surface
)
from dldna.chapter_04.utils.data import get_dataset, get_device
from dldna.chapter_04.utils.metrics import load_model
from dldna.chapter_05.visualization.gaussian_loss_surface import (
get_opt_params,
train_loss_surface,
gaussian_func # gaussian_func 추가.
)
device = get_device()
_, test_dataset = get_dataset(dataset="FashionMNIST")
small_dataset = Subset(test_dataset, torch.arange(0, 256))
data_loader = DataLoader(small_dataset, batch_size=256, shuffle=True)
loss_func = nn.CrossEntropyLoss()
act_name = "ReLU" # Tanh로 실험하려면 이 부분을 변경
model_file = f"SimpleNetwork-{act_name}.pth"
trained_model, _ = load_model(model_file=model_file, path="./tmp/opts/ReLU")
trained_model = trained_model.to(device)
top_n = 2
top_eigenvalues, top_eigenvectors = hessian_eigenvectors(
model=trained_model,
loss_func=loss_func,
data_loader=data_loader,
top_n=top_n,
is_cuda=True
)
d_min, d_max, d_num = -1, 1, 30 # 5장의 30을 사용
lambda1 = np.linspace(d_min, d_max, d_num).astype(np.float32)
lambda2 = np.linspace(d_min, d_max, d_num).astype(np.float32)
x, y, z = xy_perturb_loss(
model=trained_model,
top_eigenvectors=top_eigenvectors,
data_loader=data_loader,
loss_func=loss_func,
lambda1=lambda1,
lambda2=lambda2,
device=device # device 추가
)
# --- Optimization and Visualization ---
# Find the parameters that best fit the data.
popt, _, offset = get_opt_params(x, y, z) # offset 사용
print(f"Optimal parameters: {popt}")
# Get a new z using the optimized surface function (Gaussian).
# No need for global g_offset, we can use the returned offset.
z_fitted = gaussian_func((x, y), *popt,offset) # offset을 더해야 함.
data = [(x, y, z_fitted)] # Use z_fitted
axes = visualize_loss_surface(data, act_name=act_name, color="C0", size=6, levels=80, alpha=0.7, plot_3d=False)
ax = axes[0]
# Train with different weight decays and plot trajectories.
for n, weight_decay in enumerate([0.0, 6.0, 10.0, 18.0, 20.0]):
# for n, weight_decay in enumerate([0.0]): # For faster testing
points_sgd_m = train_loss_surface(
lambda params: optim.SGD(params, lr=0.1, momentum=0.7, weight_decay=weight_decay),
[d_min, d_max],
200,
(*popt, offset) # unpack popt and offset
)
ax.plot(
points_sgd_m[-1, 0],
points_sgd_m[-1, 1],
color=f"C{n}",
marker="o",
markersize=10,
zorder=2,
label=f"wd={weight_decay:0.1f}"
)
ax.ticklabel_format(axis='both', style='scientific', scilimits=(0, 0))
plt.legend()
plt.show()Function parameters = [ 4.59165436 0.34582255 -0.03204057 -1.09810435 1.54530407]
Optimal parameters: [ 4.59165436 0.34582255 -0.03204057 -1.09810435 1.54530407]
train_loss_surface: SGD
SGD: Iter=1 loss=4.7671 w=[-0.8065, 0.9251]
SGD: Iter=200 loss=1.9090 w=[0.3458, -0.0320]
train_loss_surface: SGD
SGD: Iter=1 loss=4.7671 w=[-0.2065, 0.3251]
SGD: Iter=200 loss=1.9952 w=[0.1327, -0.0077]
train_loss_surface: SGD
SGD: Iter=1 loss=4.7671 w=[0.1935, -0.0749]
SGD: Iter=200 loss=2.0293 w=[0.0935, -0.0051]
train_loss_surface: SGD
SGD: Iter=1 loss=4.7671 w=[0.9935, -0.8749]
SGD: Iter=200 loss=2.0641 w=[0.0587, -0.0030]
train_loss_surface: SGD
SGD: Iter=1 loss=4.7671 w=[1.1935, -1.0749]
SGD: Iter=200 loss=2.0694 w=[0.0537, -0.0027]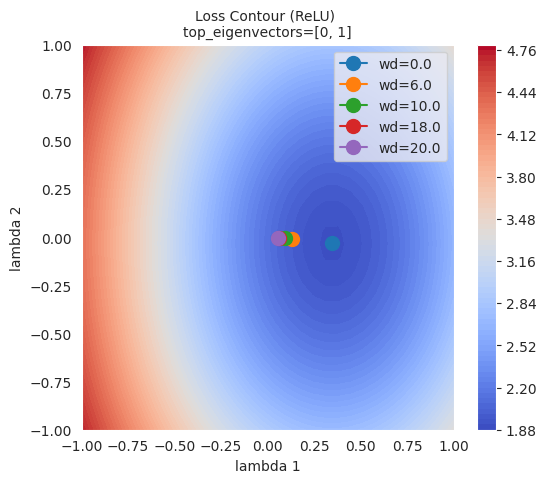
Wie in der Abbildung zu sehen ist, kann beobachtet werden, dass je größer die L2-Regulierung (Weight Decay) ist, desto weiter entfernt sich der Endpunkt, den der Optimizer erreicht hat, vom tiefsten Punkt der Verlustfunktion. Dies liegt daran, dass die L2-Regulierung verhindert, dass die Gewichte zu groß werden, um das Überanpassen des Modells zu vermeiden.
Die L1-Regulierung erstellt ein sparsames Modell (sparse model), indem sie einige Gewichte auf 0 setzt. Sie ist nützlich, wenn man die Komplexität des Modells reduzieren und unnötige Merkmale eliminieren möchte. Im Gegensatz dazu sorgt die L2-Regulierung dafür, dass alle Gewichte klein gehalten werden, aber nicht vollständig auf 0 gesetzt werden. Die L2-Regulierung zeigt im Allgemeinen eine stablilere Konvergenz und wird daher als „weiche Regulierung“ bezeichnet, da sie die Gewichte allmählich reduziert.
L1-Regulierung und L2-Regulierung werden je nach den Eigenschaften des Problems, den Daten und dem Ziel des Modells unterschiedlich angewendet. Obwohl im Allgemeinen die L2-Regulierung häufiger verwendet wird, kann es sinnvoll sein, beide Regulierungen zu testen und zu prüfen, welche bessere Leistungsresultate liefert. Darüber hinaus kann auch die Elastic Net-Regulierung, eine Kombination von L1- und L2-Regulierung, in Betracht gezogen werden.
Elastic Net ist eine Regularisierungs-Methode, die L1-Regulierung und L2-Regulierung kombiniert. Sie nutzt die Vorteile beider Regulierungen und kompensiert ihre Nachteile, um flexiblere und effektivere Modelle zu erstellen.
Kernpunkte:
Formel:
Die Kostenfunktion von Elastic Net wird wie folgt ausgedrückt:
\(Cost = Loss + \lambda_1 \sum_{i} |w_i| + \lambda_2 \sum_{i} (w_i)^2\)
Loss: Die Verlustfunktion des ursprünglichen Modells (z.B. MSE, Cross-Entropy)λ₁: Hyperparameter zur Anpassung der Stärke der L1-Regulierungλ₂: Hyperparameter zur Anpassung der Stärke der L2-Regulierungwᵢ: Gewichte des ModellsVorteile:
λ₁ und λ₂ gesteuert werden. Bei λ₁=0 wird es zur L2-Regulierung (Ridge), bei λ₂=0 zur L1-Regulierung (Lasso).Nachteile:
λ₁ und λ₂, eingestellt werden, was das Tuning komplizierter machen kann als bei L1- oder L2-Regulierung.Anwendungsfälle:
Zusammenfassung: Elastic Net ist eine leistungsfähige Regularisierungsmethode, die die Vorteile von L1 und L2 kombiniert. Obwohl es Hyperparameter-Tuning erfordert, kann es in verschiedenen Problemen gute Leistungen zeigen.
Dropout ist eine der leistungsfähigsten Regularisierungsmethoden, um Overfitting in neuronalen Netzen zu verhindern. Während des Lernprozesses werden zufällige Neuronen deaktiviert (dropout), um die Übereinstimmung bestimmter Neuronen oder Kombinationen von Neuronen mit den Trainingsdaten zu reduzieren. Dies hat eine ähnliche Wirkung wie Ensemble-Lernen, bei dem verschiedene Personen unterschiedliche Aspekte lernen und sich dann zusammenfassen, um das Problem zu lösen. Es fördert die unabhängige Lernung wichtiger Merkmale durch jedes Neuron, was die Generalisierungsfähigkeit des Modells verbessert. Im Allgemeinen wird es auf vollständig verbundene Schichten (fully connected layer) angewendet und der Deaktivierungsrate liegt zwischen 20% und 50%. Dropout wird nur während des Trainings angewendet, während bei der Inferenz alle Neuronen verwendet werden.
In PyTorch kann Dropout wie folgt einfach implementiert werden.
import torch.nn as nn
class Dropout(nn.Module):
def __init__(self, dropout_rate):
super(Dropout, self).__init__()
self.dropout_rate = dropout_rate
def forward(self, x):
if self.training:
mask = torch.bernoulli(torch.ones_like(x) * (1 - self.dropout_rate)) / (1 - self.dropout_rate)
return x * mask
else:
return x
# Usage example. Drops out 0.5 (50%).
dropout = Dropout(dropout_rate=0.5)
# Example input data
inputs = torch.randn(1000, 100)
# Forward pass (during training)
dropout.train()
outputs_train = dropout(inputs)
# Forward pass (during inference)
dropout.eval()
outputs_test = dropout(inputs)
print("Input shape:", inputs.shape)
print("Training output shape:", outputs_train.shape)
print("Test output shape", outputs_test.shape)
print("Dropout rate (should be close to 0.5):", 1 - torch.count_nonzero(outputs_train) / outputs_train.numel())Input shape: torch.Size([1000, 100])
Training output shape: torch.Size([1000, 100])
Test output shape torch.Size([1000, 100])
Dropout rate (should be close to 0.5): tensor(0.4997)Die Implementierung ist sehr einfach. Den mask-Wert mit dem Eingabetensor multiplizieren, um bestimmte Neuronen zu deaktivieren. Die Dropout-Schicht besitzt keine eigenen lernbaren Parameter und hat die einfache Aufgabe, einen Teil der Eingaben zufällig auf 0 zu setzen. In realen neuronalen Netzen wird die Dropout-Schicht zwischen anderen Schichten (z.B. lineare Schichten, Faltungsschichten) eingefügt. Während des Trainings werden Neuronen bei Dropout zufällig entfernt, während bei der Inferenz alle Neuronen verwendet werden. Dabei wird zur Angleichung der Skalierung von Ausgabewerten beim Training und bei der Inferenz die inverted dropout Methode eingesetzt. Inverted Dropout skaliert während des Trainings voraus, indem es durch (1 - dropout_rate) dividiert, sodass es bei der Inferenz ohne zusätzliche Berechnungen direkt angewendet werden kann. Dies ermöglicht es, bei der Inferenz ähnliche Effekte wie beim Ensemble-Lernen zu erzielen. Das heißt, es liefert Effekte, als würden mehrere Teilnetze (sub-networks) durchschnittlich berechnet, und steigert gleichzeitig die Berechnungseffizienz.
Um zu sehen, wie effektiv Dropout ist, betrachten wir ein einfaches Datenbeispiel in einem Diagramm. Der Quellcode befindet sich in chapter_06/plot_dropout.py, und da es sich um unwichtigen Code handelt, wird er hier nicht dargestellt. Da der Code ausführlich kommentiert ist, sollte er einfach zu verstehen sein. Das Diagramm zeigt, dass das Modell mit Dropout (blau) eine viel höhere Testgenauigkeit aufweist.
from dldna.chapter_06.plot_dropout import plot_dropout_effect
plot_dropout_effect()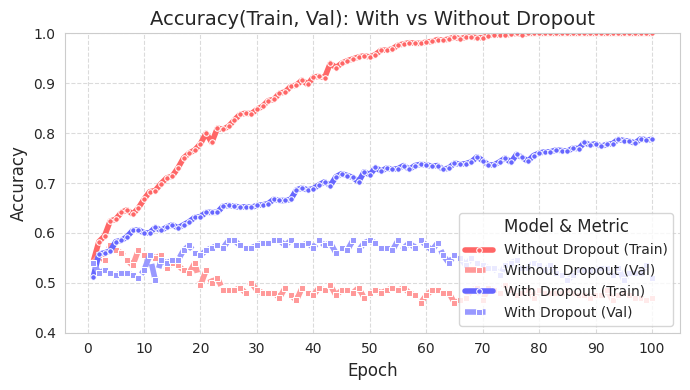
Die Trainingsgenauigkeit des Modells mit Dropout (With Dropout) ist niedriger als die des Modells ohne Dropout (Without Dropout), aber die Validierungs-genauigkeit ist höher. Dies bedeutet, dass Dropout das Overfitting auf die Trainingsdaten reduziert und die Generalisierungsfähigkeit des Modells verbessert.
Batch-Normalisierung ist eine Methode, die gleichzeitig die Rolle einer Regularisierung spielt und die Stabilität der Daten während des Trainings verbessert. Batch-Normalisierung wurde erstmals in einem Paper von Ioffe und Szegedy [Referenz 2] im Jahr 2015 vorgeschlagen. In Deep Learning ändert sich die Verteilung der Aktivierungen, wenn Daten durch jede Schicht fließen (internal covariate shift). Dies verlangsamt das Training und macht das Modell instabil (wegen der Veränderung der Verteilung werden mehr Berechnungsschritte benötigt). Je tiefer die Schichten sind, desto schlimmer wird dieses Problem. Batch-Normalisierung normalisiert die Daten in Mini-Batch-Größe, um dies zu lindern.
Das Kernkonzept der Batch-Normalisierung ist die Normalisierung der Daten in Mini-Batch-Größe. Das folgende Codebeispiel macht dies verständlich.
# Calculate the mean and variance of the mini-batch
batch_mean = x.mean(dim=0)
batch_var = x.var(dim=0, unbiased=False)
# Perform normalization
x_norm = (x - batch_mean) / torch.sqrt(batch_var + epsilon)
# Apply scale and shift parameters
y = gamma * x_norm + betaAllgemein führt die Batch-Normalisierung zu einer angemessenen Verteilungsänderung der gesamten Daten innerhalb eines Minibatches, indem sie die Varianz und den Mittelwert der Daten verwendet. Zuerst wird eine Normalisierung durchgeführt, gefolgt von der Anwendung einer bestimmten Skalierungs- und Verschiebungseingenschaft. Der oben genannte Gamma ist der Skalierungsparameter und Beta ist der Verschiebungsparameter. Es ist hilfreich, einfach \(y = ax + b\) zu betrachten. Das Epsilon, das während der Normalisierung verwendet wird, ist ein sehr kleiner Konstantwert (1e-5 oder 1e-7), der häufig in numerischer Analyse vorkommt. Dieser Wert dient der numerischen Stabilität. Die Batch-Normalisierung bietet die folgenden zusätzlichen Vorteile.
Lassen Sie uns zufällige Daten mit zwei Merkmalen erzeugen und diese sowohl in einem Fall ohne Skalierung und Verschiebung als auch in einem Fall mit angewendeten Skalierungs- und Verschiebungsparametern grafisch vergleichen. Durch die Visualisierung kann man leicht verstehen, welche numerische Bedeutung die Normalisierung innerhalb eines Minibatches hat.
import numpy as np
import seaborn as sns
import matplotlib.pyplot as plt
# Generate data
np.random.seed(42)
x = np.random.rand(50, 2) * 10
# Batch normalization (including scaling parameters)
def batch_normalize(x, epsilon=1e-5, gamma=1.0, beta=0.0):
mean = x.mean(axis=0)
var = x.var(axis=0)
x_norm = (x - mean) / np.sqrt(var + epsilon)
x_scaled = gamma * x_norm + beta
return x_norm, mean, x_scaled
# Perform normalization (gamma=1.0, beta=0.0 is pure normalization)
x_norm, mean, x_norm_scaled = batch_normalize(x, gamma=1.0, beta=0.0)
# Perform normalization and scaling (apply gamma=2.0, beta=1.0)
_, _, x_scaled = batch_normalize(x, gamma=2.0, beta=1.0)
# Set Seaborn style
sns.set_style("whitegrid")
sns.set_context("notebook", font_scale=1.2)
# Visualization
fig, (ax1, ax2, ax3) = plt.subplots(1, 3, figsize=(18, 5))
# Original data
sns.scatterplot(x=x[:, 0], y=x[:, 1], ax=ax1, color='royalblue', alpha=0.7)
ax1.scatter(mean[0], mean[1], color='red', marker='*', s=200, label='Mean')
ax1.set(title='Original Data',
xlabel='Feature 1',
ylabel='Feature 2',
xlim=(-2, 12),
ylim=(-2, 12))
ax1.legend()
# After normalization (gamma=1, beta=0)
sns.scatterplot(x=x_norm[:, 0], y=x_norm[:, 1], ax=ax2, color='crimson', alpha=0.7)
ax2.scatter(0, 0, color='blue', marker='*', s=200, label='Mean (0,0)')
ax2.axhline(y=0, color='k', linestyle='--', alpha=0.3)
ax2.axvline(x=0, color='k', linestyle='--', alpha=0.3)
ax2.set(title='After Normalization (γ=1, β=0)',
xlabel='Normalized Feature 1',
ylabel='Normalized Feature 2',
xlim=(-2, 12),
ylim=(-2, 12))
ax2.legend()
# After scaling and shifting (gamma=2, beta=1)
sns.scatterplot(x=x_scaled[:, 0], y=x_scaled[:, 1], ax=ax3, color='green', alpha=0.7)
ax3.scatter(1, 1, color='purple', marker='*', s=200, label='New Mean')
ax3.axhline(y=1, color='k', linestyle='--', alpha=0.3)
ax3.axvline(x=1, color='k', linestyle='--', alpha=0.3)
ax3.set(title='After Scale & Shift (γ=2, β=1)',
xlabel='Scaled Feature 1',
ylabel='Scaled Feature 2',
xlim=(-2, 12),
ylim=(-2, 12))
ax3.legend()
plt.tight_layout()
plt.show()
# Print statistics
print("\nOriginal Data Statistics:")
print(f"Mean: {mean}")
print(f"Variance: {x.var(axis=0)}")
print("\nNormalized Data Statistics (γ=1, β=0):")
print(f"Mean: {x_norm.mean(axis=0)}")
print(f"Variance: {x_norm.var(axis=0)}")
print("\nScaled Data Statistics (γ=2, β=1):")
print(f"Mean: {x_scaled.mean(axis=0)}")
print(f"Variance: {x_scaled.var(axis=0)}")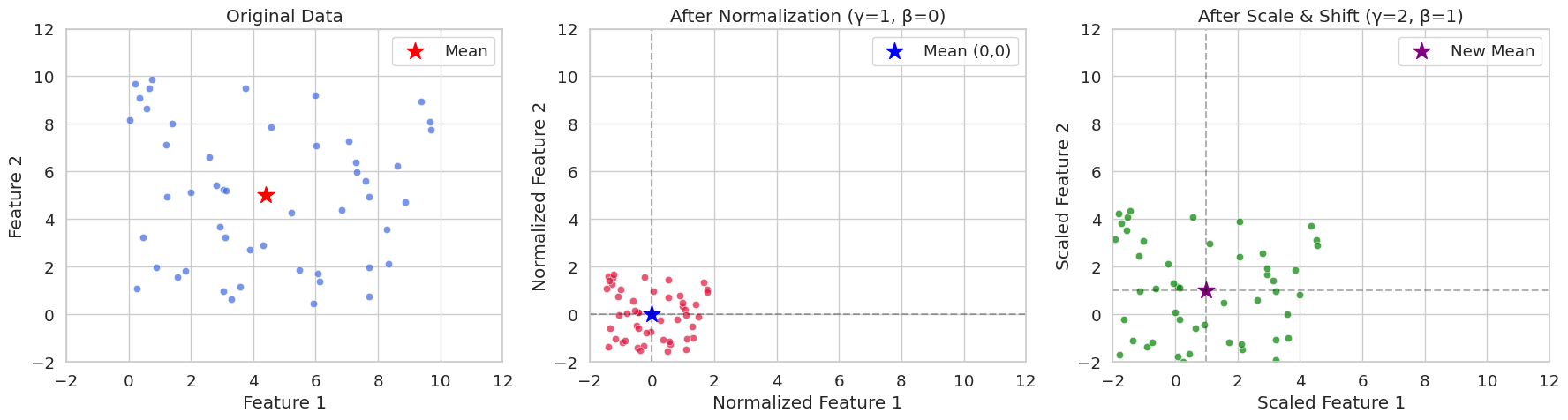
Original Data Statistics:
Mean: [4.40716778 4.99644709]
Variance: [8.89458134 8.45478364]
Normalized Data Statistics (γ=1, β=0):
Mean: [-2.70894418e-16 -3.59712260e-16]
Variance: [0.99999888 0.99999882]
Scaled Data Statistics (γ=2, β=1):
Mean: [1. 1.]
Variance: [3.9999955 3.99999527]In seed(42) kann man häufig die zufällige Initialisierung auf 42 festlegen sehen. Dies ist eine programmiererische Praxis, und es könnte auch eine andere Zahl sein. Die 42 ist die Zahl aus Douglas Adams’ Roman “Sternenfahrts-Handbuch für galaktische Hitchhiker”, wo sie als die “Antwort auf das Leben, das Universum und alles” erwähnt wird. Deshalb wird sie von Programmierern oft in Beispielcodes u.ä. als Konvention verwendet.
In PyTorch wird die Implementierung üblicherweise durch das Einfügen einer Batch-Normalisierungsschicht in den neuronalen Netzwerkschichten durchgeführt. Hier ist ein Beispiel dafür.
import torch.nn as nn
class SimpleNet(nn.Module):
def __init__(self):
super().__init__()
self.network = nn.Sequential(
nn.Linear(784, 256),
nn.BatchNorm1d(256), # 배치 정규화 층
nn.ReLU(),
nn.Linear(256, 10)
)
def forward(self, x):
return self.network(x)Die Implementierung der Batch-Normalisierung in PyTorch kann auf Basis des Original-Quellcodes wie folgt vereinfacht werden. Wie im vorherigen Kapitel, wurde dies zur Vereinfachung und Lernzwecken implementiert.
import torch
import torch.nn as nn
class BatchNorm1d(nn.Module):
def __init__(self, num_features, eps=1e-5, momentum=0.1):
super().__init__()
self.num_features = num_features
self.eps = eps
self.momentum = momentum
# Trainable parameters
self.gamma = nn.Parameter(torch.ones(num_features)) # scale
self.beta = nn.Parameter(torch.zeros(num_features)) # shift
# Running statistics to be tracked
self.register_buffer('running_mean', torch.zeros(num_features))
self.register_buffer('running_var', torch.ones(num_features))
def forward(self, x):
if self.training:
# Calculate mini-batch statistics
batch_mean = x.mean(dim=0) # Mean per channel
batch_var = x.var(dim=0, unbiased=False) # Variance per channel
# Update running statistics (important)
self.running_mean = (1 - self.momentum) * self.running_mean + self.momentum * batch_mean
self.running_var = (1 - self.momentum) * self.running_var + self.momentum * batch_var
# Normalize
x_norm = (x - batch_mean) / torch.sqrt(batch_var + self.eps)
else:
# During inference, use the stored statistics
x_norm = (x - self.running_mean) / torch.sqrt(self.running_var + self.eps)
# Apply scale and shift
return self.gamma * x_norm + self.betaDer wichtigste Unterschied zur grundlegenden Implementierung besteht im Update von Statistiken während der Ausführung. Während des Trainings werden die Statistiken (Mittelwert und Varianz) der Minibatches aufgehoben, um letztendlich den Gesamtmittelwert und die Gesamtvarianz zu ermitteln. Dies wird durch das Verfolgen der Bewegung mit einem exponentiellen gleitenden Mittelwert (Exponential Moving Average), das einen Momentum (Standardwert 0.1) verwendet, erreicht. Die während des Trainings gewonnenen Mittelwerte und Varianzen werden dann bei der Inferenz verwendet, um eine genaue Varianz und Abweichung für die Inferenzdaten anzuwenden und so Konsistenz zwischen Training und Inferenz zu garantieren.
Natürlich ist diese Implementierung stark vereinfacht für Lernzwecke. Der verwendete Code befindet sich unter (https://github.com/pytorch/pytorch/blob/main/torch/nn/modules/batchnorm.py). Die tatsächliche Implementierung von BatchNorm1d ist viel komplexer, da Frameworks wie PyTorch und TensorFlow neben der grundlegenden Logik auch CUDA-Optimierungen, Gradient-Optimierungen, verschiedene Einstellungsverarbeitungen und Integrationen mit C/C++ umfassen.
Batch Normalization (BN) hat seit seinem Vorschlag durch Ioffe & Szegedy im Jahr 2015 eine zentrale Technik im Training von Deep-Learning-Modellen geworden. BN normalisiert die Eingaben jeder Schicht, um das Lernverhalten zu beschleunigen, Gradientenverschwinden/-explosion zu mildern und einen gewissen Regulierungseffekt zu bieten. In diesem Tiefgang werden wir den Forward-Pass und Backpropagation-Prozess von BN detailliert untersuchen und dessen Effekte mathematisch analysieren.
Batch Normalization wird in Mini-Batch-Einheiten durchgeführt. Wenn die Größe des Mini-Batches \(B\) und die Dimension der Merkmale (features) \(D\) ist, kann die Eingabedatenmatrix \(\mathbf{X}\) als \(B \times D\) Matrix dargestellt werden. BN wird für jede Merkmalsdimension unabhängig durchgeführt; daher betrachten wir in dieser Erklärung nur die Berechnungen für eine einzelne Merkmalsdimension.
Berechnung des Mini-Batch-Mittels:
\(\mu_B = \frac{1}{B} \sum_{i=1}^{B} x_i\)
Hierbei steht \(x_i\) für den Wert der entsprechenden Merkmalen des \(i\)-ten Samples im Mini-Batch.
Berechnung der Mini-Batch-Varianz:
\(\sigma_B^2 = \frac{1}{B} \sum_{i=1}^{B} (x_i - \mu_B)^2\)
Normalisierung:
\(\hat{x_i} = \frac{x_i - \mu_B}{\sqrt{\sigma_B^2 + \epsilon}}\)
Hierbei ist \(\epsilon\) eine kleine Konstante, die verhindert, dass der Nenner null wird.
Skalierung und Verschiebung (Scale and Shift):
\(y_i = \gamma \hat{x_i} + \beta\)
Hier sind \(\gamma\) und \(\beta\) lernbare Parameter, die für Skalierung und Verschiebung zuständig sind. Diese Parameter dienen dazu, die Darstellungsfähigkeit der normalisierten Daten wiederherzustellen.
Die Backpropagation bei Batch Normalization besteht darin, die Gradienten der Verlustfunktion (loss function) nach jedem Parameter mithilfe der Kettenregel (chain rule) zu berechnen. Dieser Prozess kann visuell durch einen Berechnungsgraphen dargestellt werden. (Hier wird dies in vereinfachter Form als ASCII-Art gezeigt)
x_i --> [-] --> [/] --> [*] --> [+] --> y_i
| ^ ^ ^ ^
| | | | |
| | | | +---> beta
| | | +---> gamma
| | +---> sqrt(...) + epsilon
| +---> mu_B, sigma_B^2Berechnung von \(\frac{\partial \mathcal{L}}{\partial \beta}\) und \(\frac{\partial \mathcal{L}}{\partial \gamma}\):
\(\frac{\partial \mathcal{L}}{\partial \beta} = \sum_{i=1}^{B} \frac{\partial \mathcal{L}}{\partial y_i} \cdot \frac{\partial y_i}{\partial \beta} = \sum_{i=1}^{B} \frac{\partial \mathcal{L}}{\partial y_i}\)
\(\frac{\partial \mathcal{L}}{\partial \gamma} = \sum_{i=1}^{B} \frac{\partial \mathcal{L}}{\partial y_i} \cdot \frac{\partial y_i}{\partial \gamma} = \sum_{i=1}^{B} \frac{\partial \mathcal{L}}{\partial y_i} \cdot \hat{x_i}\)
Berechnung von \(\frac{\partial \mathcal{L}}{\partial \hat{x_i}}\):
\(\frac{\partial \mathcal{L}}{\partial \hat{x_i}} = \frac{\partial \mathcal{L}}{\partial y_i} \cdot \frac{\partial y_i}{\partial \hat{x_i}} = \frac{\partial \mathcal{L}}{\partial y_i} \cdot \gamma\)
Berechnung von \(\frac{\partial \mathcal{L}}{\partial \sigma_B^2}\):
\(\frac{\partial \mathcal{L}}{\partial \sigma_B^2} = \sum_{i=1}^{B} \frac{\partial \mathcal{L}}{\partial \hat{x_i}} \cdot \frac{\partial \hat{x_i}}{\partial \sigma_B^2} = \sum_{i=1}^{B} \frac{\partial \mathcal{L}}{\partial \hat{x_i}} \cdot (x_i - \mu_B) \cdot (-\frac{1}{2})(\sigma_B^2 + \epsilon)^{-3/2}\)
Berechnung von \(\frac{\partial \mathcal{L}}{\partial \mu_B}\):
\(\frac{\partial \mathcal{L}}{\partial \mu_B} = \sum_{i=1}^{B} \frac{\partial \mathcal{L}}{\partial \hat{x_i}} \cdot \frac{\partial \hat{x_i}}{\partial \mu_B} + \frac{\partial \mathcal{L}}{\partial \sigma_B^2} \cdot \frac{\partial \sigma_B^2}{\partial \mu_B} = \sum_{i=1}^{B} \frac{\partial \mathcal{L}}{\partial \hat{x_i}} \cdot \frac{-1}{\sqrt{\sigma_B^2 + \epsilon}} + \frac{\partial \mathcal{L}}{\partial \sigma_B^2} \cdot (-2)\frac{1}{B}\sum_{i=1}^B (x_i-\mu_B)\)
Da \(\sum_{i=1}^B (x_i - \mu_B) = 0\) gilt. \(\frac{\partial \mathcal{L}}{\partial \mu_B} = \sum_{i=1}^{B} \frac{\partial \mathcal{L}}{\partial \hat{x_i}} \cdot \frac{-1}{\sqrt{\sigma_B^2 + \epsilon}}\)
\(\frac{\partial \mathcal{L}}{\partial x_i}\) Berechnung:
\(\frac{\partial \mathcal{L}}{\partial x_i} = \frac{\partial \mathcal{L}}{\partial \hat{x_i}} \cdot \frac{\partial \hat{x_i}}{\partial x_i} + \frac{\partial \mathcal{L}}{\partial \mu_B} \cdot \frac{\partial \mu_B}{\partial x_i} + \frac{\partial \mathcal{L}}{\partial \sigma_B^2} \cdot \frac{\partial \sigma_B^2}{\partial x_i} = \frac{\partial \mathcal{L}}{\partial \hat{x_i}} \cdot \frac{1}{\sqrt{\sigma_B^2 + \epsilon}} + \frac{\partial \mathcal{L}}{\partial \mu_B} \cdot \frac{1}{B} + \frac{\partial \mathcal{L}}{\partial \sigma_B^2} \cdot \frac{2}{B}(x_i - \mu_B)\)
Batch Normalization verhindert, dass die Eingaben der Aktivierungsfunktion in extremen Werten liegen, indem es die Eingaben jeder Schicht normalisiert. Dies hilft dabei, das Problem des Gradientenverschwindens/-explodierenden Gradienten bei Aktivierungsfunktionen wie Sigmoid oder tanh zu mildern.
Problem des Gradientenverschwindens: Wenn die Eingaben der Aktivierungsfunktion sehr groß oder sehr klein sind, nähert sich der Gradient dieser Funktion 0 an. Während des Backpropagation-Prozesses führt dies dazu, dass der Gradient verschwindet. Batch Normalization normalisiert die Eingaben auf einen Mittelwert von 0 und eine Varianz von 1, sodass die Eingaben der Aktivierungsfunktion in einem angemessenen Bereich bleiben und das Problem des Gradientenverschwindens gemildert wird.
Problem der explodierenden Gradienten: Wenn die Eingaben der Aktivierungsfunktion sehr groß sind, kann der Gradient extrem groß werden. Batch Normalization begrenzt den Bereich der Eingaben, wodurch auch das Problem der explodierenden Gradienten gemildert wird.
Während des Trainings berechnet Batch Normalization die Mittelwerte und Varianzen auf Mini-Batch-Ebene. Bei der Inferenz sind jedoch Schätzungen für den Mittelwert und die Varianz über das gesamte Trainingsdatensatz erforderlich. Dafür berechnet Batch Normalization während des Trainingsprozesses laufendes Mittel (running mean) und laufende Varianz (running variance).
Berechnung des laufenden Mittels:
\(\text{running\_mean} = (1 - \text{momentum}) \times \text{running\_mean} + \text{momentum} \times \mu_B\)
Berechnung der laufenden Varianz:
\(\text{running\_var} = (1 - \text{momentum}) \times \text{running\_var} + \text{momentum} \times \sigma_B^2\)
Hierbei ist momentum ein Hyperparameter, der typischerweise auf 0.1 oder 0.01 gesetzt wird.
Während der Inferenz werden die während des Trainingsprozesses berechneten running_mean und running_var verwendet, um die Eingaben zu normalisieren.
Batch Normalization (BN): Verwendet Statistiken zwischen den Samples innerhalb eines Minibatches. Es ist von der Batchgröße beeinflusst und schwer anwendbar auf RNNs.
Layer Normalization (LN): Verwendet Statistiken über die Merkmalsdimensionen innerhalb jedes einzelnen Samples. Es ist nicht von der Batchgröße beeinflusst und leicht anwendbar auf RNNs.
Instance Normalization (IN): Berechnet Statistiken unabhängig für jedes Sample und jeden Kanal. Wird hauptsächlich in Bildgenerierungsaufgaben wie Style Transfer verwendet.
Group Normalization (GN): Teilt die Kanäle in Gruppen auf und berechnet Statistiken innerhalb jeder Gruppe. Es kann als Alternative zu BN verwendet werden, wenn die Batchgröße klein ist.
Jede Normalisierungstechnik hat Vor- und Nachteile in verschiedenen Situationen, daher muss je nach Problemeigenschaften und Modellarchitektur die geeignete Technik ausgewählt werden.
Die Optimierung von Hyperparametern hat einen sehr wichtigen Einfluss auf die Modellleistung. Ihre Bedeutung wurde bereits in den 1990er Jahren bekannt. In der zweiten Hälfte der 1990er Jahre wurde festgestellt, dass bei Support Vector Machines (SVM) gleiche Modelle je nach Parameter des Kernfunktion (C, gamma usw.) eine entscheidende Rolle bei der Leistung spielen. Um die Mitte der 2010er Jahre konnte bewiesen werden, dass Bayesianische Optimierung bessere Ergebnisse als manuelles Tuning liefert und wurde zu einem zentralen Bestandteil automatisierten Tunings (automated tuning) wie Google AutoML (2017) entwickelt.
Es gibt verschiedene Methoden, um Hyperparameter zu optimieren. Zu den bekanntesten gehören:
Grid Search (Gitter-Suche): Dies ist die grundlegendste Methode, bei der für jeden Hyperparameter eine Liste möglicher Werte angegeben wird und alle Kombinationen dieser Werte ausprobiert werden. Sie ist nützlich, wenn die Anzahl der Hyperparameter gering ist und das Spektrum der möglichen Werte für jeden Parameter begrenzt ist. Allerdings sind die Berechnungskosten sehr hoch, da jede Kombination getestet werden muss. Es eignet sich für einfache Modelle oder kleinere Suchräume.
Random Search (Zufallssuche): Diese Methode erzeugt Kombinationen durch zufällige Auswahl der Werte für jeden Hyperparameter und bewertet die Leistung des Modells auf Basis dieser Kombinationen. Wenn einige Hyperparameter einen größeren Einfluss auf die Leistung haben, kann dies effektiver sein als Gitter-Suche (Bergstra & Bengio, 2012).
Bayesian Optimization (Bayes’sche Optimierung): Diese Methode wählt intelligente Kombinationen von Hyperparametern auf Basis früherer Suchergebnisse und verwendet ein Wahrscheinlichkeitsmodell (in der Regel ein Gaußsches Prozess) um die nächsten zu probierenden Kombinationen auszuwählen. Die optimale Stelle wird durch Maximierung einer Erwerbsfunktion (acquisition function) bestimmt. Da sie den Suchraum für Hyperparameter effizient erkundet, kann sie mit weniger Versuchen bessere Kombinationen finden als Gitter-Suche oder Zufallssuche.
Neben diesen gibt es auch Evolutionäre Algorithmen, die auf genetischen Algorithmen basieren, und gradientenbasierte Optimierungsmethoden.
Im Folgenden wird ein Beispiel gezeigt, wie Hyperparameter eines einfachen neuronalen Netzes mit Bayes’scher Optimierung optimiert werden.
Bayes’sche Optimierung erhielt in den 2010er Jahren zunehmend an Bedeutung. Im Gegensatz zu Gitter-Suche oder Zufallssuche, die zufällige oder systematische Kombinationen ausprobieren, wählt Bayes’sche Optimierung intelligente Parameterkombinationen auf Basis früherer Versuche. Dies ist ein großer Vorteil.
Die Bayes’sche Optimierung besteht im Wesentlichen aus den folgenden drei Schritten:
init_points definiert, wird verwendet, um das Modell zu trainieren und die Leistung zu bewerten.import torch
import torch.nn as nn
import torch.optim as optim
from dldna.chapter_04.models.base import SimpleNetwork
from dldna.chapter_04.utils.data import get_data_loaders, get_device
from bayes_opt import BayesianOptimization
from dldna.chapter_04.experiments.model_training import train_model, eval_loop
def train_simple_net(hidden_layers, learning_rate, batch_size, epochs):
"""Trains a SimpleNetwork model with given hyperparameters.
Uses CIFAR100 dataset and train_model from Chapter 4.
"""
device = get_device() # Use the utility function to get device
# Get data loaders for CIFAR100
train_loader, test_loader = get_data_loaders(dataset="CIFAR100", batch_size=batch_size)
# Instantiate the model with specified activation and hidden layers.
# CIFAR100 images are 3x32x32, so the input size is 3*32*32 = 3072.
model = SimpleNetwork(act_func=nn.ReLU(), input_shape=3*32*32, hidden_shape=hidden_layers, num_labels=100).to(device)
# Optimizer: Use Adam
optimizer = optim.Adam(model.parameters(), lr=learning_rate)
# Train the model using the training function from Chapter 4
results = train_model(model, train_loader, test_loader, device, optimizer=optimizer, epochs=epochs, save_dir="./tmp/tune",
retrain=True) # retrain=True로 설정
# Return the final test accuracy
return results['test_accuracies'][-1]
def train_wrapper(learning_rate, batch_size, hidden1, hidden2):
"""Wrapper function for Bayesian optimization."""
return train_simple_net(
hidden_layers=[int(hidden1), int(hidden2)],
learning_rate=learning_rate,
batch_size=int(batch_size),
epochs=10
)
def optimize_hyperparameters():
"""Runs hyperparameter optimization."""
# Set the parameter ranges to be optimized.
pbounds = {
"learning_rate": (1e-4, 1e-2),
"batch_size": (64, 256),
"hidden1": (64, 512), # First hidden layer
"hidden2": (32, 256) # Second hidden layer
}
# Create a Bayesian optimization object.
optimizer = BayesianOptimization(
f=train_wrapper,
pbounds=pbounds,
random_state=1,
allow_duplicate_points=True
)
# Run optimization
optimizer.maximize(
init_points=4,
n_iter=10,
)
# Print the best parameters and accuracy
print("\nBest parameters found:")
print(f"Learning Rate: {optimizer.max['params']['learning_rate']:.6f}")
print(f"Batch Size: {int(optimizer.max['params']['batch_size'])}")
print(f"Hidden Layer 1: {int(optimizer.max['params']['hidden1'])}")
print(f"Hidden Layer 2: {int(optimizer.max['params']['hidden2'])}")
print(f"\nBest accuracy: {optimizer.max['target']:.4f}")
if __name__ == "__main__":
print("Starting hyperparameter optimization...")
optimize_hyperparameters()Das folgende Beispiel führt eine Hyperparameter-Optimierung mit dem Paket BayesOpt durch. Das Trainingsziel ist das SimpleNetwork (aus Kapitel 4 definiert), und der CIFAR100-Datensatz wird verwendet. Die Funktion train_wrapper dient als Zielfunktion für BayesOpt, trainiert das Modell mit den gegebenen Hyperparametern und gibt die endgültige Testgenauigkeit zurück.
pbounds gibt das Suchintervall für jeden Hyperparameter an. Bei optimizer.maximize ist init_points die Anzahl der initialen zufälligen Suchschritte, n_iter die Anzahl der Wiederholungen der bayesianischen Optimierung. Die gesamte Anzahl der Experimente beträgt daher init_points + n_iter.
Beim Durchsuchen von Hyperparametern sind folgende Punkte zu beachten:
n_iter zu erhöhen.Kürzlich hat das Framework BoTorch in der Domäne der Hyperparameter-Optimierung für Deep Learning Aufmerksamkeit erhalten. BoTorch ist ein auf PyTorch basierendes Bayesianisches Optimierungsframework, das 2019 von FAIR (Facebook AI Research, heute Meta AI) entwickelt wurde. Bayes-Opt ist eine ältere, seit 2016 entwickelte Bibliothek für Bayesianische Optimierung und bietet eine intuitive und einfache Schnittstelle (API im Stil von scikit-learn), die es weit verbreitet macht.
Die Vor- und Nachteile beider Bibliotheken sind klar:
Daher ist Bayes-Opt für einfache Probleme oder schnelle Prototypen geeignet, während BoTorch für komplexe Hyperparameter-Optimierungen von Deep-Learning-Modellen, große/hochdimensionale Probleme und fortgeschrittene Bayesianische Optimierungstechniken (z.B. multi-task, Constraint-Optimierung) empfohlen wird.
Um BoTorch zu verwenden, müssen im Gegensatz zu Bayes-Opt einige Kernkonzepte für die Anfangskonfiguration verstanden werden (Stellvertretermodelle, Normalisierung der Eingabedaten, Erwerbsfunktionen).
Stellvertreter(Surrogate) Modell:
Ein Stellvertretermodell ist ein Modell, das die tatsächliche Zielfunktion (hier die Validierungspräzision des Deep-Learning-Modells) annähert. Üblicherweise werden Gaußprozesse (GP) verwendet. GP werden anstelle der rechenintensiven tatsächlichen Zielfunktion genutzt, um schnell und kostengünstig Ergebnisse vorherzusagen. BoTorch bietet die folgenden GP-Modelle:
SingleTaskGP: Das grundlegende Gaußprozessmodell, das für Einzelzieloptimierungsprobleme geeignet ist und effektiv bei bis zu 1000 Datenpunkten arbeitet.MultiTaskGP: Für die gleichzeitige Optimierung mehrerer Zielfunktionen (multi-objective optimization) verwendet. Zum Beispiel kann sowohl die Präzision als auch die Inferenzzeit des Modells optimiert werden.SAASBO (Sparsity-Aware Adaptive Subspace Bayesian Optimization): Ein Modell, das auf hochdimensionalen Parameterräumen spezialisiert ist. Es geht davon aus, dass in hochdimensionalen Räumen Sparsität existiert und führt eine effiziente Exploration durch.Normalisierung der Eingabedaten:
Da Gaußprozesse empfindlich gegenüber der Skalierung von Daten sind, ist die Normalisierung der Eingabedaten (Hyperparameter) wichtig. Üblicherweise werden alle Hyperparameter in den Bereich [0, 1] transformiert. BoTorch bietet die Transformationen Normalize und Standardize.
Erwerbsfunktion (Acquisition Function): Erwerbfunktionen basieren auf dem Surrogatmodell (GP) und werden verwendet, um die nächste Kombination von Hyperparametern zu bestimmen, die experimentiert werden soll. Erwerbfunktionen spielen die Rolle, das Gleichgewicht zwischen “Exploration” und “Exploitation” aufrechtzuerhalten. BoTorch bietet eine Vielzahl von Erwerbfunktionen an.
ExpectedImprovement (EI): Eine der am häufigsten verwendeten Erwerbfunktionen. Sie berücksichtigt die Wahrscheinlichkeit, ein besseres Ergebnis als das aktuelle Optimum zu erzielen, und den Grad dieser Verbesserung.LogExpectedImprovement (LogEI): Die logarithmische Transformation von EI. Numerisch stabiler und sensibler gegenüber kleinen Veränderungen.UpperConfidenceBound (UCB): Eine Erwerbfunktion, die stärker auf Exploration fokussiert ist. Sie erforscht Bereiche mit hoher Unsicherheit aktivierter.ProbabilityOfImprovement (PI): Zeigt die Wahrscheinlichkeit an, das aktuelle Optimum zu verbessern.qExpectedImprovement (qEI): Auch als q-batch EI bekannt und für parallele Optimierung verwendet. Wählt mehrere Kandidaten auf einmal aus.qNoisyExpectedImprovement (qNEI): q-batch Noisy EI, verwendet in Umgebungen mit Rauschen.Der gesamte Code befindet sich in package/botorch_optimization.py. Er kann direkt von der Kommandozeile ausgeführt werden. Der vollständige Code enthält detaillierte Kommentare, daher werde ich hier nur die wichtigsten Teile jedes Codes erläutern.
def __init__(self, max_trials: int = 80, init_samples: int = 10):
self.param_bounds = torch.tensor([
[1e-4, 64.0, 32.0, 32.0], # 최소값
[1e-2, 256.0, 512.0, 512.0] # 최대값
], dtype=torch.float64)In der Initialisierungsteil werden die Minimal- und Maximalwerte für jeden Hyperparameter festgelegt. max_trials ist die Gesamtzahl der Versuche, init_samples ist die Anzahl der anfänglichen zufälligen Experimente (entspricht init_points in Bayes-Opt). init_samples wird üblicherweise auf 2-3-fach der Anzahl der Parameter gesetzt. Im obigen Beispiel gibt es 4 Hyperparameter, daher sind etwa 8-12 geeignet. Der Grund für die Verwendung von torch.float64 ist die numerische Stabilität. Bei der Bayesschen Optimierung, insbesondere beim Gaußschen Prozess, wird bei der Berechnung der Kernmatrix die Cholesky-Zerlegung verwendet, wobei float32 aufgrund von Genauigkeitsproblemen zu Fehlern führen kann.
def tune(self):
# 가우시안 프로세스 모델 학습
model = SingleTaskGP(configs, accuracies)
mll = ExactMarginalLogLikelihood(model.likelihood, model)
fit_gpytorch_mll(mll)Gaußsche Prozesse basierte Surrogatmodelle verwenden SingleTaskGP. ExactMarginalLogLikelihood ist die Verlustfunktion zum Trainieren des Modells und fit_gpytorch_mll trainiert das Modell mit dieser Verlustfunktion.
acq_func = LogExpectedImprovement(
model,
best_f=accuracies.max().item()
)Die Erwerbfunktion LogExpectedImprovement wird verwendet. Da der Logarithmus angewendet wird, ist sie numerisch stabil und reagiert empfindlich auf kleine Änderungen.
candidate, _ = optimize_acqf( # 획득 함수 최적화로 다음 실험할 파라미터 선택
acq_func, bounds=bounds, # 획득 함수와 파라미터 범위 지정
q=1, # 한 번에 하나의 설정만 선택
num_restarts=10, # 최적화 재시작 횟수
raw_samples=512 # 초기 샘플링 수
)optimize_acqf Funktion optimiert die Erwerbsfunktion, um die nächste Kombination von Hyperparametern (Kandidat) für das Experiment auszuwählen.
q=1: Wählt nur einen Kandidaten auf einmal (keine q-Batch-Optimierung).num_restarts=10: Führt die Optimierung in jedem Schritt 10-mal mit verschiedenen Startpunkten durch, um das Fallen in lokale Optima zu vermeiden.raw_samples=512: Extrahiert 512 Stichproben aus dem Gaußschen Prozess, um den Wert der Erwerbsfunktion zu schätzen.num_restarts und raw_samples haben einen großen Einfluss auf das Trade-off zwischen Exploration und Exploitation in der Bayesianischen Optimierung. num_restarts bestimmt die Gründlichkeit der Optimierung, während raw_samples die Genauigkeit der Erwerbsfunktionsschätzung beeinflusst. Je höher diese Werte sind, desto höher sind die Rechenkosten, aber auch die Wahrscheinlichkeit, bessere Ergebnisse zu erzielen. Allgemein können folgende Werte verwendet werden:
num_restarts=5, raw_samples=256num_restarts=10, raw_samples=512num_restarts=20, raw_samples=1024from dldna.chapter_06.botorch_optimizer import run_botorch_optimization
run_botorch_optimization(max_trials=80, init_samples=5)Ergebnis Datensatz : FashionMNIST Epochen : 20 Initiale Experimente : 5 Mal Wiederholte Experimente : 80 Mal
| Optimale Parameter | Bayes-Opt | Botorch |
|---|---|---|
| Lernrate | 6e-4 | 1e-4 |
| Batch-Größe | 173 | 158 |
| hid 1 | 426 | 512 |
| hid 2 | 197 | 512 |
| Genauigkeit | 0.7837 | 0.8057 |
Es ist eine einfache Vergleich, aber die Genauigkeit von BoTorch ist höher. Bei einfachen Optimierungssuchen wird Bayes-Opt empfohlen, bei fortgeschrittenen Suchen jedoch BoTorch.
Herausforderung: Wie kann die Vorhersageunsicherheit eines Modells quantifiziert werden und dazu verwendet werden, aktiv zu lernen?
Frust des Forschers: Traditionelle Deep-Learning-Modelle liefern Vorhersagergebnisse als Punktschätzungen (point estimates), aber in realen Anwendungen ist die Kenntnis der Unsicherheit einer Vorhersage von großer Bedeutung. Zum Beispiel muss ein autonomes Fahrzeug beim Vorhersagen der nächsten Position eines Fußgängers wissen, wie unsicher diese Vorhersage ist, um sicher zu fahren. Gaußsche Prozesse sind leistungsstarke Werkzeuge zur Quantifizierung der Vorhersageunsicherheit auf Basis der bayesianischen Wahrscheinlichkeitstheorie, haben aber den Nachteil, dass sie rechenaufwändig sind und schwer an große Datenmengen anzupassen.
Gaußsche Prozesse (Gaussian Process, GP) sind zentrale Modelle im Bayesianischen Maschinellen Lernen, die unsichere Vorhersagen (uncertainty-aware prediction) bereitstellen. Wir haben bereits kurz auf die Verwendung von Gaußschen Prozessen als Surrogatmodelle in der bayesianischen Optimierung eingegangen; hier wollen wir uns detaillierter mit den grundlegenden Prinzipien und der Bedeutung von Gaußschen Prozessen selbst befassen.
Ein GP wird als “Wahrscheinlichkeitsverteilung über Mengen von Funktionswerten” definiert. Im Gegensatz zu deterministischen Funktionen wie \(y = f(x)\), die eine einzelne Ausgabe für einen gegebenen Eingang liefern, vorhersagt ein GP nicht eine einzelne Ausgabe für eine gegebene Eingabe \(x\), sondern eine Verteilung möglicher Ausgaben. Zum Beispiel anstatt festzulegen, “die höchste Temperatur morgen wird 25 Grad sein”, kann man sagen, “es gibt eine 95%-ige Wahrscheinlichkeit, dass die höchste Temperatur morgen zwischen 23 und 27 Grad liegt”. Wenn man Fahrrad fährt, um nach Hause zu kommen, ist der grobe Weg bereits festgelegt, aber jedes Mal wird der tatsächliche Weg unterschiedlich sein. In solchen Fällen sind unsichere Vorhersagen erforderlich.
Die mathematischen Werkzeuge zur Behandlung unsicherer Vorhersagen basieren auf dem von dem 19. Jahrhundert Mathematiker Gauß vorgeschlagenen Normalverteilung (Gaußsche Verteilung). Darauf basierend entwickelten sich in den 1940er Jahren die GP. Es war die Zeit des Zweiten Weltkrieges, als Wissenschaftler mit unsicheren Daten umgehen mussten wie Radar-Signalverarbeitung, Entschlüsselung und Wetterdaten-Verarbeitung. Ein berühmtes Beispiel ist das von Norbert Wiener entwickelte Prädiktionsfilter für Flugabwehrgeschütze. In der Zeitreihenanalyse arbeitete Harald Cramér, in der Wahrscheinlichkeitstheorie Andrey Kolmogorov an den mathematischen Grundlagen der GP. 1951 prägte Daniel Krige mit der Anwendung von GP zur Vorhersage von Erzlagerstätten die praktische Anwendung. In den 1970er Jahren wurden durch Statistiker räumliche Anwendungen, Designs für Computerexperimente und Bayesianische Optimierung im maschinellen Lernen systematisiert. Heute spielen sie in fast allen Bereichen, die mit Unsicherheiten umgehen müssen, wie Künstlicher Intelligenz, Robotik und Klimavorhersage eine zentrale Rolle. Insbesondere in der Tiefen Lernung sind tief gehende Kernel-GP durch Meta-Lernen hervorragend aufgetreten, insbesondere in Bereichen wie der Vorhersage molekularer Eigenschaften.
Heute werden Gaußsche Prozesse in verschiedenen Bereichen eingesetzt, darunter Künstliche Intelligenz, Robotik und Klimamodellierung. Insbesondere in der Tiefen Lernung zeigen sie durch Meta-Lernen und tiefgehende Kernel-GP in Bereichen wie der Vorhersage molekularer Eigenschaften hervorragende Leistungen.
Der Gaussian Process (GP) ist ein stochastisches Modell, das auf Kernmethoden (kernel methods) basiert und in Regressions- und Klassifikationsproblemen weit verbreitet ist. GPs definieren eine Verteilung über Funktionen selbst, was es ermöglicht, die Unsicherheit von Vorhersagen zu quantifizieren. In diesem Deep Dive untersuchen wir detailliert die mathematischen Grundlagen des Gaussian Processes, beginnend mit der multivariaten Normalverteilung bis hin zur Perspektive stochastischer Prozesse und erforschen verschiedene Anwendungen im maschinellen Lernen.
Der erste Schritt zum Verstehen von Gaussian Processes ist das Verständnis der multivariaten Normalverteilung. Ein \(d\)-dimensionaler Zufallsvektor \(\mathbf{x} = (x_1, x_2, ..., x_d)^T\), der multivariat normalverteilt ist, hat die folgende Wahrscheinlichkeitsdichtefunktion (probability density function).
\(p(\mathbf{x}) = \frac{1}{(2\pi)^{d/2}|\mathbf{\Sigma}|^{1/2}} \exp\left(-\frac{1}{2}(\mathbf{x} - \boldsymbol{\mu})^T \mathbf{\Sigma}^{-1} (\mathbf{x} - \boldsymbol{\mu})\right)\)
Dabei ist \(\boldsymbol{\mu} \in \mathbb{R}^d\) der Mittelwertsvektor und \(\mathbf{\Sigma} \in \mathbb{R}^{d \times d}\) die Kovarianzmatrix (covariance matrix). Die Kovarianzmatrix muss positiv definit sein.
Kernmerkmale:
Lineare Transformation: Eine lineare Transformation einer multivariat normalverteilten Zufallsvariablen ist ebenfalls multivariat normalverteilt. Das heißt, wenn \(\mathbf{x} \sim \mathcal{N}(\boldsymbol{\mu}, \mathbf{\Sigma})\) und \(\mathbf{y} = \mathbf{A}\mathbf{x} + \mathbf{b}\), dann ist \(\mathbf{y} \sim \mathcal{N}(\mathbf{A}\boldsymbol{\mu} + \mathbf{b}, \mathbf{A}\mathbf{\Sigma}\mathbf{A}^T)\).
Bedingte Verteilung (Conditional Distribution): Die bedingte Verteilung einer multivariaten Normalverteilung ist ebenfalls normalverteilt. Wenn \(\mathbf{x}\) in \(\mathbf{x} = (\mathbf{x}_1, \mathbf{x}_2)^T\) unterteilt wird und der Mittelwert und die Kovarianzmatrix wie folgt unterteilt sind:
\(\boldsymbol{\mu} = \begin{pmatrix} \boldsymbol{\mu}_1 \\ \boldsymbol{\mu}_2 \end{pmatrix}, \quad \mathbf{\Sigma} = \begin{pmatrix} \mathbf{\Sigma}_{11} & \mathbf{\Sigma}_{12} \\ \mathbf{\Sigma}_{21} & \mathbf{\Sigma}_{22} \end{pmatrix}\)
dann ist die bedingte Verteilung von \(\mathbf{x}_2\) gegeben \(\mathbf{x}_1\) wie folgt:
\(p(\mathbf{x}_2 | \mathbf{x}_1) = \mathcal{N}(\boldsymbol{\mu}_{2|1}, \mathbf{\Sigma}_{2|1})\)
\(\boldsymbol{\mu}_{2|1} = \boldsymbol{\mu}_2 + \mathbf{\Sigma}_{21}\mathbf{\Sigma}_{11}^{-1}(\mathbf{x}_1 - \boldsymbol{\mu}_1)\) \(\mathbf{\Sigma}_{2|1} = \mathbf{\Sigma}_{22} - \mathbf{\Sigma}_{21}\mathbf{\Sigma}_{11}^{-1}\mathbf{\Sigma}_{12}\)
Randverteilung (Marginal Distribution): Die Randverteilungen einer multivariaten Normalverteilung sind ebenfalls normalverteilt. In der obigen Zerlegung ist die Randverteilung von \(\mathbf{x}_1\) wie folgt. \(p(\mathbf{x}_1) = \mathcal{N}(\boldsymbol{\mu_1}, \mathbf{\Sigma}_{11})\)
Ein Gaußscher Prozess ist eine Wahrscheinlichkeitsverteilung über Funktionen. Das bedeutet, dass ein gegebener Funktion \(f(x)\) einem Gaußschen Prozess folgt, wenn der Vektor der Funktionswerte \((f(x_1), f(x_2), ..., f(x_n))^T\) für jede endliche Menge von Eingabepunkten \(\{x_1, x_2, ..., x_n\}\) einer multivariaten Normalverteilung folgt.
Definition: Ein Gaußscher Prozess ist durch eine Mittelfunktion (mean function) \(m(x)\) und eine Kovarianzfunktion (covariance function oder Kernel-Funktion) \(k(x, x')\) definiert.
\(f(x) \sim \mathcal{GP}(m(x), k(x, x'))\)
Sichtweise als stochastischer Prozess (Stochastic Process): Ein Gaußscher Prozess ist eine Art von stochastischem Prozess, bei dem jeder Element eines Index-Satzes (hier der Eingaberaum) einer Zufallsvariable zugeordnet wird. Im Falle eines Gaußschen Prozesses bilden diese Zufallsvariablen eine gemeinsame Normalverteilung.
Die Kernel-Funktion ist ein wesentlicher Bestandteil des Gaußschen Prozesses. Sie repräsentiert die Ähnlichkeit zwischen zwei Eingaben \(x\) und \(x'\) und bestimmt die Eigenschaften des Gaußschen Prozesses.
Wichtige Rollen:
Verschiedene Kernel-Funktionen:
RBF (Radial Basis Function) Kernel (oder Squared Exponential-Kernel):
\(k(x, x') = \sigma^2 \exp\left(-\frac{\|x - x'\|^2}{2l^2}\right)\)
Matérn-Kernel:
\(k(x, x') = \sigma^2 \frac{2^{1-\nu}}{\Gamma(\nu)}\left(\sqrt{2\nu}\frac{\|x - x'\|}{l}\right)^\nu K_\nu\left(\sqrt{2\nu}\frac{\|x - x'\|}{l}\right)\)
\(\nu\): Weichheitsparameter (smoothness parameter)
\(K_\nu\): Modifizierte Bessel-Funktion (modified Bessel function)
\(\nu = 1/2, 3/2, 5/2\) werden häufig als Halbzahlen (half-integer) verwendet.
Je größer \(\nu\), desto ähnlicher wird der RBF-Kernel.
Periodischer (Periodic) Kernel:
\(k(x, x') = \sigma^2 \exp\left(-\frac{2\sin^2(\pi|x-x'|/p)}{l^2}\right)\)
Linearer (Linear) Kernel:
\(k(x,x') = \sigma_b^2 + \sigma_v^2(x - c)(x' -c)\)
Regression:
Die Gaußsche-Prozess-Regression (Gaussian Process Regression) befasst sich damit, für eine neue Eingabe \(\mathbf{x}_*\) die Ausgabe \(f(\mathbf{x}_*)\) basierend auf gegebenen Trainingsdaten \(\mathcal{D} = \{(\mathbf{x}_i, y_i)\}_{i=1}^n\) vorherzusagen. Die Vorhersageverteilung (predictive distribution) wird durch Kombination der a-priori-Verteilung (prior distribution) des Gaußschen Prozesses mit den Trainingsdaten berechnet.
Klassifikation:
Die Gaußsche-Prozess-Klassifikation (Gaussian Process Classification) modelliert die latente Funktion \(f(\mathbf{x})\) als Gaußscher Prozess und definiert die Klassifikationswahrscheinlichkeiten durch diese latente Funktion. Zum Beispiel wird in binären Klassifikationsproblemen die latente-Funktionswerte über eine Logit- oder Probitfunktion in Wahrscheinlichkeiten umgewandelt.
Da in Klassifikationsproblemen die a-posteriori-Verteilung nicht in geschlossener Form vorliegt, werden approximative Inferenzmethoden wie die Laplace-Approximation oder variationelle Inferenz verwendet. ### 5. Vor- und Nachteile von Gaußschen Prozessen sowie Vergleich mit tiefem Lernen
Vorteile:
Nachteile:
Vergleich mit tiefem Lernen:
In jüngster Zeit werden auch Modelle erforscht, die tiefes Lernen und Gaußsche Prozesse kombinieren (z. B. Deep Kernel Learning).
Im Allgemeinen denken wir an eine Funktion als eine einzelne Linie, aber ein Gaußscher Prozess betrachtet sie als “Menge möglicher Linien”. Mathematisch ausgedrückt ist es wie folgt.
\(f(t) \sim \mathcal{GP}(m(t), k(t,t'))\)
Wenn wir das Beispiel eines Fahrradstandorts nehmen, dann stellt \(m(t)\) die Mittelfunktion dar und “vorausgesagt wird, dass der Pfad ungefähr so verlaufen wird”. \(k(t,t')\) ist die Kovarianzfunktion (oder Kernel), welche angibt, “wie stark sich Standorte zu verschiedenen Zeitpunkten voneinander abhängen?”. Es gibt mehrere typische Kernels. Ein häufig verwendeter Kernel ist der RBF (Radial Basis Function).
\(k(t,t') = \sigma^2 \exp\left(-\frac{(t-t')^2}{2l^2}\right)\)
Diese Formel ist sehr intuitiv. Je näher die beiden Zeitpunkte \(t\) und \(t'\) beieinander liegen, desto größer ist der Wert, und je weiter sie auseinanderliegen, desto kleiner ist der Wert. Es ähnelt “wenn man den aktuellen Standort kennt, kann man den Standort in ein paar Minuten etwa vorhersagen, aber den Standort in ferner Zukunft nicht so gut”.
Nehmen wir an, der Kernel (\(K\)) ist RBF und betrachten wir ein Beispiel. Stellen Sie sich vor, Sie betreiben einen Fahrrad-Sharing-Dienst (oder auch autonomes Fahren). Wir schätzen die gesamte Bewegungspfad eines Fahrrads basierend nur auf wenigen GPS-Datenpunkten.
Grundlegende Formel der Vorhersage
\(f_* | X, y, X_* \sim \mathcal{N}(\mu_*, \Sigma_*)\)
Diese Formel bedeutet “basierend auf unseren GPS-Aufzeichnungen (\(X\), \(y\)), folgt die Position des Fahrrads zu unbekannten Zeitpunkten (\(X_*\)) einer Normalverteilung mit Mittelwert \(\mu_*\) und Unsicherheit \(\Sigma_*\)”.
Berechnung der Standortvorhersage
\(\mu_* = K_*K^{-1}y\)
Diese Formel zeigt, wie die Position des Fahrrads vorhergesagt wird. \(K_*\) repräsentiert den ‘zeitlichen Zusammenhang’ zwischen den zu prognostizierenden Zeitpunkten und den GPS-Aufzeichnungspunkten; \(K^{-1}\) ist die Inverse der Matrix, die die Beziehungen zwischen den vorhandenen Datenpunkten darstellt; \(y\) sind die GPS-Aufzeichnungen. Je dichter die Daten liegen, desto größer wird \(K_*\) und desto mehr benachbarte Daten können in Betracht gezogen werden.
Um dies mit einem realen Szenario zu erklären: 1. Zunächst geht man davon aus, dass das Fahrrad überall hinfahren könnte (\(K_{**}\) ist groß) 2. Je mehr GPS-Aufzeichnungen vorhanden sind (\(K_*\) wird größer) 3. Und je konsistentere Aufzeichnungen vorliegen (\(K^{-1}\) stabil) 4. Desto geringer wird die Unsicherheit der Standortvorhersage
Effekt der Formeln auf Daten Die unsicheren Vorhersagen hängen von der Menge an GPS-Daten ab und sehen wie folgt aus: 1. Abschnitt mit häufigen GPS-Aufzeichnungen: geringe Unsicherheit - \(K_*\) ist groß und es gibt viele Daten, so dass \(K_*K^{-1}K_*^T\) groß ist - Daher wird \(\Sigma_*\) klein und der Pfadschätzwert genau 2. Abschnitt mit seltenen GPS-Aufzeichnungen: hohe Unsicherheit - \(K_*\) ist klein und es gibt wenige Daten, so dass \(K_*K^{-1}K_*^T\) klein ist - Daher wird \(\Sigma_*\) groß und die Unsicherheit der Pfadvorhersage hoch
Einfach gesagt, wird \(K\) größer, je dichter die Zeitintervall-Daten liegen, und daher wird die Unsicherheit geringer.
Lassen Sie uns das Beispiel des Fahrradpfads verstehen, um es zu veranschaulichen.
import numpy as np
import seaborn as sns
import matplotlib.pyplot as plt
# 시각화 스타일 설정
sns.set_style("whitegrid")
plt.rcParams['font.size'] = 10
# 데이터셋 1: 5개 관측점
time1 = np.array([0, 2, 5, 8, 10]).reshape(-1, 1)
position1 = np.array([0, 2, 3, 1, 4])
# 데이터셋 2: 8개 관측점
time2 = np.array([0, 1, 2, 4, 5, 6, 8, 10]).reshape(-1, 1)
position2 = np.array([0, 1, 2.5, 1.5, 3, 2, 1, 4]) # 더 큰 변동성 추가
# 예측할 시간점 생성: 0~10분 구간을 100개로 분할
time_pred = np.linspace(0, 10, 100).reshape(-1, 1)
# RBF 커널 함수 정의
def kernel(T1, T2, l=2.0):
sqdist = np.sum(T1**2, 1).reshape(-1, 1) + np.sum(T2**2, 1) - 2 * np.dot(T1, T2.T)
return np.exp(-0.5 * sqdist / l**2)
# 가우시안 프로세스 예측 함수
def predict_gp(time, position, time_pred):
K = kernel(time, time)
K_star = kernel(time_pred, time)
K_star_star = kernel(time_pred, time_pred)
mu_star = K_star.dot(np.linalg.inv(K)).dot(position)
sigma_star = K_star_star - K_star.dot(np.linalg.inv(K)).dot(K_star.T)
return mu_star, sigma_star
# 두 데이터셋에 대한 예측 수행
mu1, sigma1 = predict_gp(time1, position1, time_pred)
mu2, sigma2 = predict_gp(time2, position2, time_pred)
# 2개의 서브플롯 생성
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(16, 4))
# 첫 번째 그래프 (5개 데이터)
ax1.fill_between(time_pred.flatten(),
mu1 - 2*np.sqrt(np.diag(sigma1)),
mu1 + 2*np.sqrt(np.diag(sigma1)),
color='blue', alpha=0.2, label='95% confidence interval')
ax1.plot(time_pred, mu1, 'b-', linewidth=1.5, label='Predicted path')
ax1.plot(time1, position1, 'ro', markersize=6, label='GPS records')
ax1.set_xlabel('Time (min)')
ax1.set_ylabel('Position (km)')
ax1.set_title('Route Estimation (5 GPS points)')
ax1.legend(fontsize=8)
# 두 번째 그래프 (8개 데이터)
ax2.fill_between(time_pred.flatten(),
mu2 - 2*np.sqrt(np.diag(sigma2)),
mu2 + 2*np.sqrt(np.diag(sigma2)),
color='blue', alpha=0.2, label='95% confidence interval')
ax2.plot(time_pred, mu2, 'b-', linewidth=1.5, label='Predicted path')
ax2.plot(time2, position2, 'ro', markersize=6, label='GPS records')
ax2.set_xlabel('Time (min)')
ax2.set_ylabel('Position (km)')
ax2.set_title('Route Estimation (8 GPS points)')
ax2.legend(fontsize=8)
plt.tight_layout()
plt.show()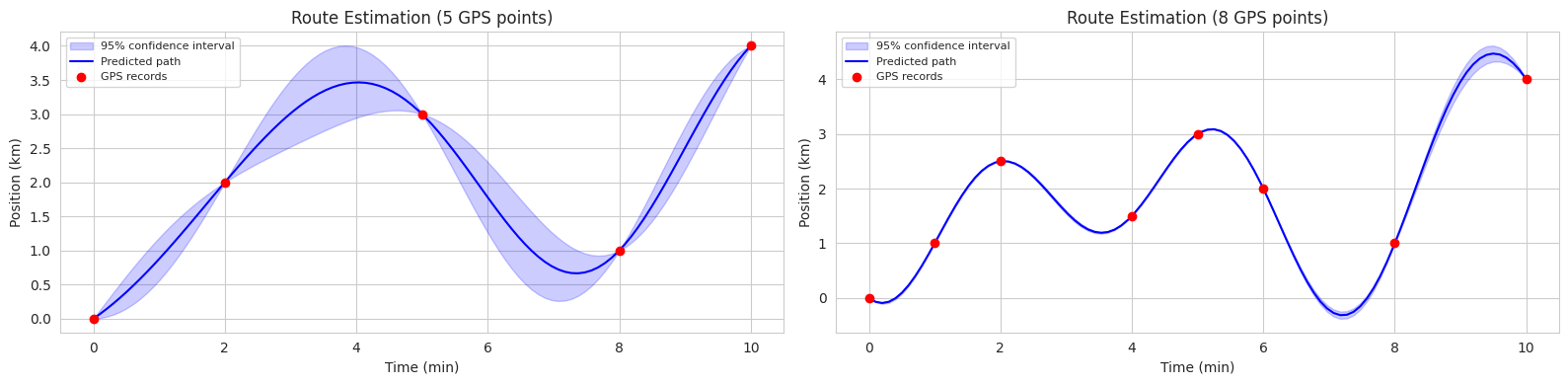
Der folgende Code ist ein Beispiel für die Schätzung von Fahrradstrecken unter Verwendung von GPs in zwei Szenarien (5 Beobachtungspunkte, 8 Beobachtungspunkte). In jedem Diagramm stellt die durchgezogene blaue Linie den vorhergesagten mittleren Pfad dar, während der blau schattierte Bereich das 95%ige Konfidenzintervall zeigt.
Auf diese Weise bieten GP nicht nur die Vorhersagergebnisse, sondern auch die Unsicherheit dieser Vorhersagen. Dies macht sie in verschiedenen Bereichen (z.B. autonomes Fahren, Roboterkontrolle, medizinische Diagnostik), in denen Unsicherheiten im Entscheidungsprozess berücksichtigt werden müssen, nützlich.
Gaußsche Prozesse werden in verschiedenen wissenschaftlichen und ingenieurtechnischen Bereichen angewendet, wie Robotersteuerung, Optimierung von Sensornetzen, Vorhersage von Molekülstrukturen, Klimamodellierung und Analyse von Astronomiedaten. Ein repräsentatives Anwendungsbeispiel im Maschinelles Lernen ist die Hyperparameter-Optimierung, wie bereits besprochen. Ein weiteres wichtiger Bereich, in dem unsichere Vorhersagen erforderlich sind, sind autonom fahrende Fahrzeuge. Hier wird das zukünftige Positionieren von anderen Fahrzeugen vorhergesagt, und bei hohen Unsicherheiten wird eine vorsichtigere Fahrt durchgeführt. Darüber hinaus werden sie in der Medizin zur Vorhersage von Patientenzuständen und im Finanzmarkt zur Vorhersage von Aktienkursen und zum Managen von Risiken aufgrund von Unsicherheit angewendet. In jüngster Zeit wird die Anwendung von GP in Bereichen wie Verstärkungslernen (Reinforcement Learning), Kombination mit Generativen Modellen im Deep Learning, Kausalinferenz und Metakernen intensiv erforscht.
Der wichtigste Bestandteil von Gaußschen Prozessen ist das Kernmodell (Covarianzfunktion). Deep Learning hat die Stärke, Darstellungen aus Daten zu lernen. Es liegt nahe, eine effektive Kombination der Vorhersagefähigkeiten von GP und den Lernfähigkeiten von tiefen neuronalen Netzen in Bezug auf Darstellung als Forschungsrichtung anzustreben. Ein repräsentatives Verfahren ist das Tiefes Kernlernverfahren (Deep Kernel Learning, DKL), bei dem anstelle eines vordefinierten Kerns wie des RBF-Kerns ein neuronales Netz verwendet wird, um direkt aus den Daten zu lernen.
Die allgemeine Struktur von DKL sieht folgendermaßen aus:
DKL wird in verschiedenen Bereichen angewendet. * Bildklassifikation (Image Classification): Verwenden von CNNs, um Bildmerkmale zu extrahieren und GP zur Durchführung der Klassifizierung. * Graphenklassifikation (Graph Classification): Verwenden von Graph Neural Networks (GNN), um Merkmale aus der Graphenstruktur zu extrahieren und GP zur Graphenklassifizierung. * Moleküleigenschaftsvorhersage (Molecular Property Prediction): Empfangen von Molekülgraphen als Eingabe, um die Eigenschaften des Moleküls (z. B. Löslichkeit, Toxizität) vorherzusagen. * Zeitreihenvorhersage (Time Series Forecasting): Verwenden von RNNS, um Merkmale aus Zeitreihendaten zu extrahieren und GP zur Vorhersage zukünftiger Werte. Hier führen wir ein einfaches Beispiel für DKL durch und werden in Teil 2 detailliertere Inhalte und Anwendungsbeispiele behandeln.
Tiefgang-Kern-Netzwerke
Zuerst definieren wir tiefgangige Kernnetzwerke. Ein Kernnetzwerk ist ein neuronales Netz, das eine Kernfunktion lernt. Dieses Netzwerk nimmt Eingabedaten auf und gibt eine Merkmalsdarstellung aus. Diese Merkmalsdarstellung wird zur Berechnung der Kernmatrix verwendet.
import torch
import torch.nn as nn
import torch.optim as optim
from torch.distributions import Normal
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
# Set seed for reproducibility
torch.manual_seed(42)
np.random.seed(42)
# Define a neural network to learn the kernel
class DeepKernel(nn.Module):
def __init__(self, input_dim, hidden_dim, output_dim):
super(DeepKernel, self).__init__()
self.fc1 = nn.Linear(input_dim, hidden_dim)
self.fc2 = nn.Linear(hidden_dim, hidden_dim)
self.fc3 = nn.Linear(hidden_dim, output_dim)
self.activation = nn.ReLU()
def forward(self, x):
x = self.activation(self.fc1(x))
x = self.activation(self.fc2(x))
x = self.fc3(x) # No activation on the final layer
return xDie Eingabe eines tiefen Kernels Neuronalen Netzes ist in der Regel ein 2D-Tensor, wobei die erste Dimension die Batch-Größe und die zweite Dimension die Dimension der Eingangsdaten ist. Die Ausgabe ist ein 2D-Tensor mit der Form (Batch-Größe, Dimensionsgröße der Merkmalsdarstellung).
GP-Schichtdefinition
Die GP-Schicht nimmt die Ausgabe des tiefen Kernel-Netzwerks entgegen, berechnet die Kernel-Matrix und bestimmt die Vorhersageverteilung.
import torch
import torch.nn as nn
# Define the Gaussian Process layer
class GaussianProcessLayer(nn.Module):
def __init__(self, num_dim, num_data):
super(GaussianProcessLayer, self).__init__()
self.num_dim = num_dim
self.num_data = num_data
self.lengthscale = nn.Parameter(torch.ones(num_dim)) # Length-scale for each dimension
self.noise_var = nn.Parameter(torch.ones(1)) # Noise variance
self.outputscale = nn.Parameter(torch.ones(1)) # Output scale
def forward(self, x, y):
# Calculate the kernel matrix (using RBF kernel)
dist_matrix = torch.cdist(x, x) # Pairwise distances between inputs
kernel_matrix = self.outputscale * torch.exp(-0.5 * dist_matrix**2 / self.lengthscale**2)
kernel_matrix += self.noise_var * torch.eye(self.num_data)
# Calculate the predictive distribution (using Cholesky decomposition)
L = torch.linalg.cholesky(kernel_matrix)
alpha = torch.cholesky_solve(y.unsqueeze(-1), L) # Add unsqueeze for correct shape
predictive_mean = torch.matmul(kernel_matrix, alpha).squeeze(-1) # Remove extra dimension
v = torch.linalg.solve_triangular(L, kernel_matrix, upper=False)
predictive_var = kernel_matrix - torch.matmul(v.T, v)
return predictive_mean, predictive_var
return predictive_mean, predictive_varDie Eingabe der GP-Schicht ist ein 2D-Tensor mit der Form (Batch-Größe, Dimension der Merkmalsdarstellung). Die Ausgabe ist ein Tupel, das die vorhergesagten Mittelwerte und Varianzen enthält. Für die Berechnung der Kernmatrix wird der RBF-Kern verwendet, und um die Berechnungseffizienz zu erhöhen, wird die Cholesky-Zerlegung zur Berechnung der Vorhersageverteilung genutzt. y.unsqueeze(-1) und .squeeze(-1) dienen dazu, die Dimensionen zwischen y und der Kernmatrix abzugleichen.
# 데이터를 생성
x = np.linspace(-10, 10, 100)
y = np.sin(x) + 0.1 * np.random.randn(100)
# 데이터를 텐서로 변환
x_tensor = torch.tensor(x, dtype=torch.float32).unsqueeze(-1) # (100,) -> (100, 1)
y_tensor = torch.tensor(y, dtype=torch.float32) # (100,)
# 딥 커널과 GP 레이어를 초기화
deep_kernel = DeepKernel(input_dim=1, hidden_dim=50, output_dim=1) # output_dim=1로 수정
gp_layer = GaussianProcessLayer(num_dim=1, num_data=len(x))
# 손실 함수와 최적화기를 정의
loss_fn = nn.MSELoss() # Use MSE loss
optimizer = optim.Adam(list(deep_kernel.parameters()) + list(gp_layer.parameters()), lr=0.01)
num_epochs = 100
# 모델을 학습
for epoch in range(num_epochs):
optimizer.zero_grad()
kernel_output = deep_kernel(x_tensor)
predictive_mean, _ = gp_layer(kernel_output, y_tensor) # predictive_var는 사용 안함
loss = loss_fn(predictive_mean, y_tensor) # Use predictive_mean here
loss.backward()
optimizer.step()
if(epoch % 10 == 0):
print(f'Epoch {epoch+1}, Loss: {loss.item()}')
# 예측을 수행
with torch.no_grad():
kernel_output = deep_kernel(x_tensor)
predictive_mean, predictive_var = gp_layer(kernel_output, y_tensor)
# 결과를 시각화
sns.set()
plt.figure(figsize=(10, 6))
plt.plot(x, y, 'bo', label='Training Data')
plt.plot(x, predictive_mean.numpy(), 'r-', label='Predictive Mean')
plt.fill_between(x, predictive_mean.numpy() - 1.96 * np.sqrt(predictive_var.numpy().diagonal()),
predictive_mean.numpy() + 1.96 * np.sqrt(predictive_var.numpy().diagonal()),
alpha=0.2, label='95% CI')
plt.legend()
plt.show()Epoch 1, Loss: 4.3467857893837725e-13
Epoch 11, Loss: 3.1288711313699757e-13
Epoch 21, Loss: 3.9212150236903054e-13
Epoch 31, Loss: 4.184870765894244e-13
Epoch 41, Loss: 2.9785689973499396e-13
Epoch 51, Loss: 3.8607078688482344e-13
Epoch 61, Loss: 3.9107123572454383e-13
Epoch 71, Loss: 2.359286811054462e-13
Epoch 81, Loss: 3.4729958167147024e-13
Epoch 91, Loss: 2.7600995490886793e-13/tmp/ipykernel_1408185/2425174321.py:40: RuntimeWarning: invalid value encountered in sqrt
plt.fill_between(x, predictive_mean.numpy() - 1.96 * np.sqrt(predictive_var.numpy().diagonal()),
/tmp/ipykernel_1408185/2425174321.py:41: RuntimeWarning: invalid value encountered in sqrt
predictive_mean.numpy() + 1.96 * np.sqrt(predictive_var.numpy().diagonal()),
Das Modelltraining verwendet die Mean Squared Error (MSE) Verlustfunktion und den Adam-Optimierer, um die Parameter des tiefen Kernnetzes und der GP-Schicht simultan zu lernen.
Das vorangegangene Beispiel zeigt die grundlegende Idee des Deep Kernel Learning (DKL). Es verwendet ein Deep-Learning-Modell (DeepKernel Klasse), um Merkmale der Eingangsdaten zu extrahieren, und diese Merkmale werden dann verwendet, um den Kern eines Gaußschen Prozesses (GP) zu berechnen. Anschließend wird GP verwendet, um den Mittelwert und die Varianz (Unsicherheit) der Vorhersage zu berechnen. Auf diese Weise kombiniert DKL die Fähigkeiten des Deep Learnings zur Merkmalsrepräsentation mit der Unsicherheitsquantifizierung von GPs, um auch bei komplexen Daten verlässliche Vorhersagen zu ermöglichen.
Möglichkeiten des DKL:
Grenzen des DKL:
Dieses Kapitel stellt verschiedene Techniken vor, die auf unterschiedliche Weise das Problem des Overfitting ansprechen:
custom_loss-Lambda-Veränderung:
lambda: Der Einfluss des Regularisierungsterms wird größer. Die Gewichte werden kleiner und das Modell einfacher, was die Wahrscheinlichkeit einer Unteranpassung erhöht.lambda: Der Einfluss des Regularisierungsterms wird geringer. Die Gewichte werden größer und das Modell komplexer, was die Wahrscheinlichkeit einer Überanpassung erhöht.Polynomielle Regression: (Code weggelassen) Eine zu hohe Ordnung führt zu Überanpassung, eine zu niedrige zu Unteranpassung.
L1/L2-Regularisierung: (Code weggelassen) Je stärker die Regularisierungsstärke (lambda) ist, desto kleiner werden die Gewichte und desto größer sind die Leistungsänderungen.
Dropout-Rate: (Code weggelassen) Eine angemessene Dropout-Rate kann Überanpassung verhindern und die Leistung verbessern. Zu hohe Raten können zu Unteranpassung führen.
Batch-Normalisierung: (Code weggelassen) Die Hinzufügung von Batch-Normalisierung beschleunigt das Lernen und fördert eine stabile Konvergenz.
Lagrange-Multiplikatorenmethode:
Backpropagation bei Batch-Normalisierung: (Herleitung weggelassen) Batch-Normalisierung normiert die Eingaben jeder Schicht, um Probleme mit verschwindenden oder explodierenden Gradienten zu mildern und das Lernen zu stabilisieren.
Visualisierung der Verlustebene: (Code weggelassen) L1-Regularisierung erzeugt rautenförmige, L2-Regularisierung kreisförmige Restriktionsbedingungen, sodass die optimalen Lösungen an verschiedenen Positionen entstehen.
Dropout-Ensemble: Dropout hat den Effekt, dass das Modell bei jedem Trainingsdurchgang mit unterschiedlichen Netzwerkstrukturen trainiert wird, ähnlich wie Ensemble-Lernen. Bei der Vorhersage werden alle Neuronen verwendet (ohne Dropout), um eine durchschnittliche Vorhersage zu treffen. Durch Monte-Carlo-Dropout kann die Unsicherheit der Vorhersagen geschätzt werden.
Methoden zur Hyperparameter-Optimierung:
Implementierung der bayesianischen Optimierung: (Code weggelassen) Verwendet ein Stellvertretermodell (z.B. Gaußscher Prozess) und eine Erwerbfunktion (z.B. Expected Improvement).
Gaußscher Prozess: Eine Wahrscheinlichkeitsverteilung über Funktionen. Definiert die Kovarianz zwischen Funktionswerten mit Hilfe einer Kernfunktion. Berechnet auf Basis der gegebenen Daten eine posteriore Verteilung, um den Mittelwert und die Varianz (Unsicherheit) der Vorhersage zu liefern.
Bedingungen für Kernfunktionen: Sie müssen positiv semidefinit sein. Die durch beliebige Eingangspunkte erzeugten Kernmatrizen (Gram-Matrix) müssen positiv semidefinite Matrizen sein. Der RBF-Kern erfüllt diese Bedingung. (Beweis weggelassen)
Erwerbfunktionen: In der bayesianischen Optimierung werden sie verwendet, um den nächsten Suchpunkt zu bestimmen. Expected Improvement (EI) berücksichtigt sowohl die Wahrscheinlichkeit als auch das Ausmaß einer Verbesserung gegenüber dem bisherigen Optimum, um den nächsten Suchpunkt auszuwählen. (Herleitung der Formeln weggelassen)
Original text:
번역:
Das ist der zu übersetzende Text.